Seroquel 25mg, 50mg, 100mg, 200mg, 300mg Quetiapine 使用法、副作用および投与量。 オンライン薬局の価格。 処方箋不要のジェネリック医薬品。
セロクエル 100mg とは何ですか? どのように使用されますか?
セロクエル 50mg は、統合失調症、双極 I 型障害、躁病の症状を治療するために使用される処方薬です。双極性障害、うつ病のエピソード;双極 I 型障害、メンテナンス;および大うつ病性障害。セロクエル 50mg は、単独で使用することも、他の薬と併用することもできます。
セロクエルは、第 2 世代の抗精神病薬と呼ばれるクラスの薬に属しています。抗躁剤。
セロクエル 200mg が 12 歳未満の子供に安全で効果的かどうかは不明です。
セロクエル 50mg の副作用の可能性は何ですか?
セロクエルは、次のような重大な副作用を引き起こす可能性があります。
- 顔の制御されていない筋肉の動き (噛む、唇を叩く、顔をしかめる、舌の動き、まばたきまたは目の動き)、
- 顔のマスクのような外観、
- 嚥下障害、
- スピーチの問題、
- 立ちくらみ、
- 重度の便秘、
- 痛みや排尿困難、
- ぼやけた視界、
- 視野狭窄、
- 眼の痛み、
- ライトの周りにハローが見える
- 非常に硬い(硬い)筋肉、
- 高熱、
- 発汗、
- 錯乱、
- 速いまたは不均一な心拍、
- 震え、
- 失神、
- 喉の渇きが増し、
- 排尿の増加、
- 口渇、
- フルーティーな口臭、
- 熱、
- 寒気、
- 口内炎、
- 皮膚のただれ、
- 喉の痛み、
- 咳、そして
- 呼吸困難
上記の症状がある場合は、すぐに医療機関を受診してください。
セロクエルの最も一般的な副作用は次のとおりです。
- スピーチの問題、
- めまい、
- 眠気、
- 疲れ、
- エネルギーの欠乏、
- 速い心拍、
- 鼻づまり、
- 食欲増進、
- 体重の増加、
- 胃のむかつき、
- 嘔吐、
- 便秘、
- ドライマウスと
- 肝機能異常検査
気になる副作用や治らない副作用がある場合は、医師に相談してください。
これらは、セロクエルの考えられるすべての副作用ではありません。詳細については、医師または薬剤師にお尋ねください。
副作用に関する医学的アドバイスについては、医師に連絡してください。 1-800-FDA-1088 で副作用を FDA に報告できます。
警告
認知症関連精神病の高齢患者の死亡率の増加;および自殺念慮と行動
認知症関連精神病の高齢患者における死亡率の増加
抗精神病薬で治療されている認知症関連精神病の高齢患者は、死亡のリスクが高くなります [警告と注意事項を参照]。セロクエルは、認知症関連の精神病患者の治療には承認されていません [警告と注意事項を参照]。
自殺念慮と行動
抗うつ薬は、短期間の研究で、子供、青年、および若年成人の自殺念慮および自殺行動のリスクを増加させました。これらの研究では、24 歳以上の患者における抗うつ薬の使用による自殺念慮および自殺行動のリスクの増加は示されませんでした。 65歳以上の患者では、抗うつ薬の使用によりリスクが低下しました[警告と注意事項を参照]。
抗うつ薬治療を開始したすべての年齢の患者では、悪化、および自殺念慮や自殺行動の出現を綿密に監視します。処方者との綿密な観察とコミュニケーションの必要性を家族と介護者に助言する[警告と注意事項を参照]。
セロクエル 50mg は、10 歳未満の小児患者への使用は承認されていません [特定集団での使用を参照]。
説明
SEROQUEL® (フマル酸クエチアピン) は、ジベンゾチアゼピン誘導体の化学クラスに属する向精神薬です。化学名は、2-[2-(4-ジベンゾ[b,f][1,4]チアゼピン-11-イル-1-ピペラジニル)エトキシ]-エタノール フマレート (2:1) (塩) です。フマル酸塩として錠剤に含まれています。すべての用量と錠剤強度は、フマル酸塩ではなく、塩基のミリグラムとして表されます。その分子式は C42H50N6O4S2•C4H4O4 で、分子量は 883.11 (フマル酸塩) です。構造式は次のとおりです。
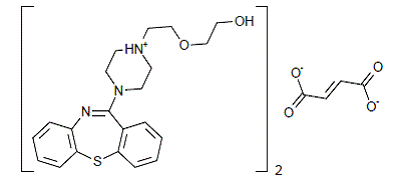
フマル酸クエチアピンは、水に適度に溶ける白色からオフホワイトの結晶性粉末です。
セロクエルは、経口投与用として、25mg(丸、桃)、50mg(丸、白)、100mg(丸、黄)、200mg(丸、白)、300mg(カプセル形、白)、 400mg(カプセル状、黄色)の錠剤。
不活性成分は、ポビドン、二塩基性リン酸二カルシウム二水和物、微結晶性セルロース、デンプングリコール酸ナトリウム、ラクトース一水和物、ステアリン酸マグネシウム、ヒプロメロース、ポリエチレングリコール、および二酸化チタンです。
25 mg 錠剤には赤色三二酸化鉄と黄色三二酸化鉄が含まれており、100 mg と 400 mg 錠剤には黄色三二酸化鉄のみが含まれています。
適応症
統合失調症
セロクエル XR は統合失調症の治療を適応としています。統合失調症におけるセロクエル XR の有効性は、統合失調症の成人を対象とした 1 回の 6 週間および 1 回の維持試験で確立されました。有効性は、統合失調症の成人を対象とした 3 件の 6 週間試験と、セロクエルで治療された統合失調症の青少年 (13~17 歳) を対象とした 1 件の 6 週間試験によって裏付けられた [参照 臨床研究 ]。
双極性障害
セロクエル 25mg XR は、双極 I 型障害に関連する躁病エピソードまたは混合エピソードの急性期治療に、単剤療法およびリチウムまたはジバルプロエックスの補助療法として適応されます。双極 I 型障害の躁病エピソードまたは混合エピソードにおけるセロクエル XR の有効性は、双極 I 障害に関連する躁病エピソードまたは混合エピソードのある成人を対象とした 3 週間の試験で確立されました。有効性は、双極 I 型障害に関連する躁病エピソードを持つ成人における 2 つの 12 週間の単剤療法試験と 1 つの 3 週間の補助的試験、および関連する躁病エピソードを持つ小児および青年 (10 ~ 17 歳) における 1 つの 3 週間の単剤療法試験によって裏付けられました。セロクエルで治療された双極 I 型障害 [参照 臨床研究 ]。
セロクエル 100mg XR は、双極性障害に伴う抑うつエピソードの急性期治療に適応されます。セロクエル XR の有効性は、双極 I または II 障害の成人を対象とした 1 件の 8 週間試験で確立され、セロクエルで治療された双極 I または II 障害の成人を対象とした 2 件の 8 週間試験で裏付けられた [参照 臨床研究 ]。
セロクエル 100mg XR は、双極 I 型障害の維持治療に、リチウムまたはジバルプロエックスの補助として適応されます。有効性は、セロクエルで治療された双極 I 型障害の成人における 2 つの維持試験から外挿されました。双極 I 型障害の維持療法に対する単剤療法の有効性は、対照臨床試験で体系的に評価されていない [参照 臨床研究 ]。
大うつ病性障害(MDD)の補助治療
セロクエル 100mg XR は、MDD の治療のための抗うつ薬の補助療法としての使用が適応とされています。 MDD における抗うつ薬の補助療法としてのセロクエル XR の有効性は、抗うつ薬治療に対する反応が不十分な MDD の成人を対象とした 2 つの 6 週間試験で確立されました [ 臨床研究 ]。
小児統合失調症および双極 I 型障害の治療における特別な考慮事項
小児統合失調症と双極 I 型障害は重篤な精神障害ですが、診断が困難な場合があります。小児統合失調症の場合、症状のプロファイルはさまざまであり、双極 I 型障害の場合、患者は躁病または混合症状の周期性のさまざまなパターンを示すことがあります。小児の統合失調症および双極 I 型障害の薬物療法は、徹底的な診断評価が行われ、薬物療法に伴うリスクが慎重に考慮された後にのみ開始することをお勧めします。小児統合失調症と双極 I 型障害の薬物治療は、心理的、教育的、社会的介入を含む総合治療プログラムの一部として適応されます。
投薬と管理
重要な管理手順
セロクエル XR 錠は、割ったり、噛んだり、砕いたりせずに、丸ごと飲み込んでください。
セロクエル 50mg XR は、食事なしまたは軽食(約 300 カロリー)と一緒に摂取することをお勧めします。 臨床薬理学 ]。
セロクエル 25mg XR は 1 日 1 回、できれば夕方に投与する必要があります。
推奨用量
承認された各適応症に対するセロクエル 50mg XR の推奨初回用量、用量設定、用量範囲、最大用量を以下の表 1 に示します。初回投与後、患者の臨床反応と忍容性に応じて、必要に応じて上方または下方に調整することができます[参照 臨床研究 ]。
統合失調症および双極Ⅰ型障害の維持療法
維持治療
患者は定期的に再評価され、維持治療の必要性とそのような治療の適切な用量を決定する必要があります[参照 臨床研究 ]。
高齢患者における用量変更
高齢者や衰弱した患者、または低血圧反応の素因がある患者では、用量漸増の速度を遅くし、目標用量を低くすることを考慮する必要があります。 特定の集団での使用 、 と 臨床薬理学 ]。必要に応じて、これらの患者では用量漸増を慎重に行う必要があります。
高齢患者は、セロクエル XR 50 mg/日から開始する必要があり、個々の患者の臨床反応と忍容性に応じて、用量を 50 mg/日ずつ増やすことができます。
肝障害患者における用量変更
肝機能障害のある患者は、セロクエル XR 50 mg/日を開始する必要があります。用量は、患者の臨床反応および忍容性に応じて、1 日 50 mg 単位で有効用量まで毎日増加させることができます。
CYP3A4阻害剤と併用する場合の用量変更
セロクエル XR の用量は、強力な CYP3A4 阻害剤(例、ケトコナゾール、イトラコナゾール、インジナビル、リトナビル、ネファゾドンなど)と併用する場合、元の用量の 6 分の 1 に減らす必要があります。 CYP3A4 阻害剤を中止する場合、セロクエル XR の用量を 6 倍に増量する必要があります [ 臨床薬理学 と 薬物相互作用 ]。
CYP3A4 インデューサーと併用する場合の用量変更
セロクエル XR の用量は、強力な CYP3A4 誘導物質(例、フェニトイン、カルバマゼピン、リファンピン、アバシミブ、Stジョンズワートなど)。用量は、個々の患者の臨床反応と耐性に基づいて滴定する必要があります。 CYP3A4 誘導剤が中止された場合、セロクエル 100mg XR の用量は 7~14 日以内に元のレベルまで減量する必要があります [ 臨床薬理学 と 薬物相互作用 ]。
以前に中止された患者の治療の再開
治療の再開に具体的に対処するデータはありませんが、セロクエル 50mg XR を 1 週間以上中止していた患者の治療を再開する場合は、最初の投与スケジュールに従うことが推奨されます。セロクエル 300mg XR を 1 週間未満休薬していた患者を再開する場合、段階的な増量は必要なく、維持量を再開することができます。
セロクエル 50mg 錠からセロクエル 100mg XR 錠への患者の切り替え
現在セロクエル(即時放出製剤)で治療されている患者は、セロクエル XR に切り替えて、1 日 1 回の総投与量と同等の用量を使用することができます。個々の投与量の調整が必要な場合があります。
抗精神病薬からの切り替え
他の抗精神病薬からセロクエル 200mg XR への患者の切り替え、または他の抗精神病薬との併用投与に関して具体的に対処するための体系的に収集されたデータはありません。以前の抗精神病薬治療の即時中止が許容される患者もいれば、より段階的な中止が最も適切な患者もいます。いずれの場合も、重複する抗精神病薬の投与期間は最小限に抑える必要があります。デポ型抗精神病薬から患者を切り替える場合、医学的に適切であれば、次に予定されている注射の代わりにセロクエル XR 療法を開始します。既存の錐体外路症候群の治療を継続する必要性は、定期的に再評価する必要があります。
供給方法
剤形と強度
- 50 mg 徐放性錠剤は、片面に「XR 50」、もう片面に無地の桃色、フィルム コーティング、カプセル形状、両凸、陰刻のある錠剤です。
- 150 mg 徐放性錠剤は、白色、フィルム コーティング、カプセル形状、両凸、片面に「XR 150」、反対面に無地の刻印のある錠剤です。
- 200 mg 徐放性錠剤は、黄色、フィルム コーティング、カプセル形状、両凸、片面に「XR 200」、反対面に無地の刻印のある錠剤です。
- 300 mg 徐放性錠剤は、淡黄色、フィルム コーティング、カプセル形状、両凸、片面に「XR 300」、反対面に無地の刻印のある錠剤です。
- 400 mg 徐放性錠剤は、白色、フィルム コーティング、カプセル形状、両凸、片面に「XR 400」、もう片面に無地の刻印のある錠剤です。
保管と取り扱い
50mg錠( NDC 0310-0280-60) ピーチ、フィルムコーティング、カプセル形状、両凸、片面に「XR 50」、もう片面に無地の刻印入り錠剤で、60 錠入りのボトルで提供されます。
150mg錠( NDC 0310-0281-60) 白色、フィルムコーティング、カプセル型、両凸、片面に「XR 150」、もう片面に無地の刻印入りの錠剤で、60 錠入りのボトルで提供されます。
200mg錠( NDC 0310-0282-60) 片面に「XR 200」、もう片面に無地の、黄色、フィルムコーティング、カプセル形状、両凸、凹みのある錠剤で、60 錠入りのボトルで提供されます。
300mg錠( NDC 0310-0283-60) 淡黄色、フィルムコーティング、カプセル形状、両凸、片面に「XR 300」、もう片面には無地の刻印入りの錠剤で、60 錠入りのボトルで提供されます。
400mg錠( NDC 0310-0284-60) 片面に「XR 400」、もう片面に無地の、白色、フィルムコーティング、カプセル型、両凸型、凹みのある錠剤で、60 錠入りのボトルで提供されます。
セロクエル XR は 25°C (77°F) で保管してください。 15 ~ 30°C (59 ~ 86°F) まで許容されるエクスカーション [参照 USP ]。
販売元: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850. 改訂: 2020 年 3 月
副作用
次の副作用については、ラベルの他のセクションで詳しく説明しています。
- 認知症に関連した精神病の高齢患者の死亡率の増加 [参照 警告と注意事項 ]
- 青年および若年成人における自殺念慮および自殺行動[参照 警告と注意事項 ]
- 認知症関連精神病の高齢患者における脳卒中を含む脳血管有害反応[参照 警告と注意事項 ]
- 神経弛緩性悪性症候群(NMS)[参照 警告と注意事項 ]
- 代謝の変化(高血糖、脂質異常症、体重増加)[参照 警告と注意事項 ]
- 遅発性ジスキネジア 警告と注意事項 ]
- 低血圧 [参照 警告と注意事項 ]
- 滝 [参照 警告と注意事項 ]
- 血圧の上昇(小児および青年)[参照 警告と注意事項 ]
- 白血球減少症、好中球減少症および無顆粒球症[参照 警告と注意事項 ]
- 白内障 [参照 警告と注意事項 ]
- QT延長[参照 警告と注意事項 ]
- 発作 [参照 警告と注意事項 ]
- 甲状腺機能低下症 [参照 警告と注意事項 ]
- 高プロラクチン血症 [参照 警告と注意事項 ]
- 認知および運動障害の可能性 [参照 警告と注意事項 ]
- 体温調節[参照] 警告と注意事項 ]
- 嚥下障害 [参照 警告と注意事項 ]
- 中断症候群 [参照 警告と注意事項 ]
- 抗コリン作用(抗ムスカリン作用)[参照 警告と注意事項 ]
臨床研究経験
臨床試験はさまざまな条件下で実施されるため、ある医薬品の臨床試験で観察された副作用の発生率を別の医薬品の臨床試験で観察された発生率と直接比較することはできず、実際に観察された発生率を反映していない可能性があります。
大人
以下の情報は、4,300 人以上の患者からなるセロクエル 200mg の臨床試験データベースから得られたものです。このデータベースには、双極性うつ病の治療のためにセロクエルに暴露された 698 人の患者、急性双極性躁病の治療のためにセロクエル 200mg に暴露された 405 人の患者 (単剤療法および補助療法)、双極 I 型障害の維持治療のためにセロクエルに補助的に暴露された 646 人の患者が含まれます。治療、および統合失調症の治療のためにセロクエル 200mg の 1 回以上の投与にさらされた約 2,600 人の患者および/または健常者。
この約 4,300 人の被験者のうち、約 4,000 人 (統合失調症 2,300 人、急性双極性躁病 405 人、双極性うつ病 698 人、双極 I 型障害の維持治療 646 人) が複数回投与効果試験に参加した患者であり、その経験は約 2400 患者年。セロクエル 200mg による治療の条件と期間は大きく異なり、(重複するカテゴリーに) 非盲検および二重盲検段階の研究、入院患者と外来患者、固定用量および用量漸増研究、および短期または長期の研究が含まれていました。暴露。有害反応は、有害反応、身体検査の結果、バイタルサイン、体重、実験室分析、心電図、および眼科検査の結果を収集することによって評価されました。
記載されている副作用の頻度は、記載されている種類の副作用を少なくとも 1 回経験した個人の割合を表しています。
短期間のプラセボ対照試験における治療中止に伴う有害反応
統合失調症
全体として、対照試験のプールでは、副作用による中止の発生率にほとんど差がありませんでした(セロクエルで 4% 対 プラセボで 3%)。しかし、傾眠(セロクエル 100mg 0.8% 対 プラセボ 0%)および低血圧(セロクエル 200mg 0.4% 対 プラセボ 0%)による中止は、薬物関連とみなされた[ 警告と注意事項 ]。
双極性障害
マニア
全体として、副作用による中止はセロクエル 25mg で 5.7%、単剤療法でプラセボで 5.1%、セロクエルで 3.6%、補助療法でプラセボで 5.9% でした。
うつ
全体として、副作用による中止は、セロクエル 300 mg で 12.3%、セロクエル 600 mg で 19.0%、プラセボで 5.2% でした。
短期のプラセボ対照試験でよく観察される有害反応
統合失調症(最大 6 週間)および双極性躁病(最大 12 週間)の急性治療試験で、セロクエル単剤療法の使用に関連して最も一般的に観察された副作用(発生率 5% 以上)は、セロクエルはプラセボの少なくとも 2 倍で、傾眠 (18%)、めまい (11%)、口渇 (9%)、便秘 (8%)、ALT 上昇 (5%)、体重増加 (5%)、消化不良 ( 5%)。
短期間のプラセボ対照試験におけるセロクエル治療患者の 2% 以上の発生率で発生する有害反応
処方者は、患者の特徴やその他の要因が臨床試験で一般的だったものとは異なる通常の医療行為における副作用の発生率を予測するために、表および表の数値を使用できないことに注意する必要があります。同様に、引用された頻度は、異なる治療法、使用法、および研究者を含む他の臨床研究から得られた数値と比較することはできません.しかし、引用された数値は、調査対象集団における副作用発生率に対する薬物因子と非薬物因子の相対的な寄与を推定するための根拠を処方医に提供します。
表 9 は、統合失調症 (最長 6 週間) および双極性躁病 (最長 12 週間) の急性治療中にセロクエルで治療された患者の 2% 以上 (用量範囲セロクエルで治療された患者の発生率は、プラセボで治療された患者の発生率よりも高かった.
双極性躁病の急性補助療法(最長 3 週間)試験で、セロクエルの使用に関連して最も一般的に観察された有害反応(発生率 5% 以上)は、セロクエルで少なくともプラセボの 2 倍の割合で観察されました。傾眠(34%)、口渇(19%)、無力症(10%)、便秘(10%)、腹痛(7%)、姿勢性低血圧(7%)、咽頭炎(6%)、体重増加(6%) %)。
表 10 は、セロクエル (100 から 800 mg/日の範囲の用量) で治療された患者の 2% 以上で、急性躁病の治療中 (最大 3 週間) に発生した副作用の発生率を、最も近いパーセントに四捨五入して列挙しています。セロクエルで治療された患者の発生率がプラセボで治療された患者の発生率よりも高かった場合、リチウムとジバルプロエックスの補助療法として。
双極性うつ病の研究 (最大 8 週間) で、セロクエルの使用に関連して最も一般的に観察された有害反応 (発生率 5% 以上) およびセロクエル 200mg で少なくともプラセボの 2 倍の割合で観察された副作用は傾眠 (57% )、口渇(44%)、めまい(18%)、便秘(10%)、無気力(5%).
表 11 は、セロクエル (300 および 600 mg/日の用量) で治療された患者の 2% 以上で双極性うつ病の治療 (最大 8 週間) 中に発生した副作用の発生率を、最も近いパーセントに四捨五入して列挙しています。セロクエルで治療された患者の発生率は、プラセボで治療された患者の発生率よりも高かった。
性別、年齢、および人種に基づく相互作用の調査では、これらの人口統計学的要因に基づく有害反応の発生に臨床的に意味のある違いは明らかになりませんでした。
短期間のプラセボ対照試験における有害反応の用量依存性
用量関連の有害反応
セロクエルの 5 つの固定用量 (75 mg、150 mg、300 mg、600 mg、および 750 mg/日) をプラセボと比較した統合失調症の研究から自然に誘発された副作用データを、副作用の用量関連性について調査しました。ロジスティック回帰分析により、次の有害反応に対する正の用量反応 (p
クエチアピンの臨床試験における有害反応で、ラベルの他の場所に記載されていないもの:
以下の有害反応もクエチアピンで報告されています:悪夢、過敏症、および血清クレアチンホスホキナーゼの上昇(NMSとは関係ありません)、乳汁漏出、徐脈(治療の開始時または開始近くに発生し、低血圧および/または失神と関連している可能性があります) ) 血小板減少、夢遊病 (およびその他の関連イベント)、γ-GT レベルの上昇、低体温症、呼吸困難、好酸球増加症、尿閉、腸閉塞および持続勃起症。
錐体外路症状 (EPS)
ジストニア
クラス効果
ジストニアの症状、筋肉群の長期にわたる異常な収縮は、治療の最初の数日間に影響を受けやすい個人に発生する可能性があります.ジストニアの症状には、首の筋肉のけいれん、時には喉の圧迫感、嚥下困難、呼吸困難、および/または舌の突出が含まれます.これらの症状は低用量でも発生する可能性がありますが、第 1 世代の抗精神病薬の用量が多いほど、より頻繁に発生し、重症度が高くなります。急性ジストニアのリスクの上昇は、男性および若い年齢層で観察されます。
EPS を測定するために 4 つの方法が使用されました: (1) パーキンソニズムとアカシジアを評価する Simpson-Angus 合計スコア (ベースラインからの平均変化)、(2) バーンズ アカシジア評価尺度 (BARS) グローバル アセスメント スコア、(3) 自発的な不調の発生率EPS(アカシジア、無動症、歯車の硬直、錐体外路症候群、筋緊張亢進、運動機能低下、首の硬直、振戦)、および(4)EPSを治療するための抗コリン薬の使用。
大人
セロクエルの 5 つの固定用量 (75、150、300、600、750 mg/日) を比較した統合失調症の 6 週間臨床試験のデータは、錐体外路症状 (EPS) の欠如と、セロクエルに関連する EPS の用量関連性の証拠を提供しました。 100mg治療。 EPS を測定するために 3 つの方法が使用されました: (1) パーキンソニズムとアカシジアを評価する Simpson-Angus 合計スコア (ベースラインからの平均変化)、(2) EPS の自発的な愁訴の発生率 (アカシジア、無動症、歯車の硬直、錐体外路症候群、筋緊張亢進、運動機能低下症、頸部硬直、および振戦)、および (3) EPS に対する抗コリン薬の使用。
表 12 では、ジストニー イベントには、項部硬直、筋緊張亢進、ジストニア、筋硬直、眼球形成が含まれていました。パーキンソニズムには、歯車のこわばり、振戦、よだれ、運動機能低下が含まれます。アカシジアには、アカシジア、精神運動性激越が含まれます。ジスキネジアイベントには、遅発性ジスキネジア、ジスキネジア、舞踏病アテトーシスが含まれます。その他の錐体外路イベントには、落ち着きのなさ、錐体外路障害、運動障害が含まれていました。
プラセボと 5 つの固定用量 (75、150、300、600、750 mg/日) の Simpson-Angus 合計スコアによって測定されたパーキンソニズムの発生率は、-0.6 でした。 -1.0、-1.2; -1.6; -1.8、および -1.8。プラセボと 5 つの固定用量の EPS を治療するための抗コリン薬の使用率は、14% でした。 11%; 10%; 8%; 12%、11%。
可変用量のセロクエルを使用した 6 つの追加のプラセボ対照臨床試験 (急性躁病で 3 つ、統合失調症で 3 つ) では、Simpson-Angus の合計スコアで評価したように、EPS の発生率にセロクエルとプラセボ治療群の間に差はありませんでした。 EPS の自発的な愁訴と、EPS を治療するための併用抗コリン薬の使用。
セロクエル 100 mg 300 mg と 600 mg を使用した双極性うつ病の治療に関する 2 つのプラセボ対照臨床試験では、EPS に関連する可能性のある副作用の発生率は、両用量群で 12%、プラセボ群で 6% でした。これらの研究では、個々の有害反応(アカシジア、錐体外路障害、振戦、ジスキネジア、ジストニア、落ち着きのなさ、不随意の筋収縮、精神運動亢進、および筋硬直)の発生率は一般的に低く、どの治療群でも 4% を超えませんでした。
つの治療グループは、治療終了時の SAS 合計スコアと BARS グローバル評価スコアの平均変化が類似していました。併用抗コリン薬の使用はまれであり、3 つの治療群で同様でした。
子供と青年
以下の情報は、1,000 人以上の小児患者からなるセロクエル 200mg の臨床試験データベースから得られたものです。このデータベースには、統合失調症の治療のためにセロクエル 200mg に暴露された 677 人の患者と、急性双極性躁病の治療のためにセロクエルに暴露された 393 人の小児および青年 (10~17 歳) が含まれています。
短期間のプラセボ対照試験における治療中止に伴う有害反応
統合失調症
クエチアピン治療患者とプラセボ治療患者の副作用による中止の発生率は、それぞれ 8.2% と 2.7% でした。セロクエル投与患者の 1% 以上で中止に至り、プラセボよりも発生率が高かった有害事象は傾眠でした (プラセボでは 2.7%、0%)。
バイポーラ I マニア
クエチアピンで治療された患者とプラセボで治療された患者の副作用による中止の発生率は、それぞれ 11.4% と 4.4% でした。セロクエル群の患者の 2% 以上で中止に至り、プラセボよりも発生率が高かった副作用は、眠気 (4.1% 対 1.1%) と疲労 (2.1% 対 0) でした。
短期のプラセボ対照試験でよく観察される有害反応
統合失調症の治療(最大 6 週間)で、青年期のクエチアピンの使用に関連して最も一般的に観察された有害反応(発生率 5% 以上、クエチアピンの発生率は少なくともプラセボの 2 倍)は、傾眠(34%)、めまいでした。 (12%)、口渇 (7%)、頻脈 (7%)。
双極性躁病治療(最長3週間)において、小児および青年におけるクエチアピンの使用に関連して最も一般的に観察された有害反応(5%以上の発生率およびプラセボの少なくとも2倍のクエチアピン発生率)は傾眠(53%)でした。めまい(18%)、疲労(11%)、食欲増進(9%)、吐き気(8%)、嘔吐(8%)、頻脈(7%)、口渇(7%)、体重増加(6%) )。
有効性が確立されていない双極性うつ病の小児および青年(10~17 歳)を対象としたセロクエル 100mg XR の急性(8 週間)試験では、セロクエル XR の使用に関連して最も一般的に観察された副作用(発生率プラセボの 2 倍以上の 5% 以上) は、めまい 7%、下痢 5%、疲労 5%、吐き気 5% でした。
短期間のプラセボ対照試験で、セロクエル 300mg 治療患者で 2% 以上の頻度で発生した有害反応
統合失調症(思春期、13~17歳)
以下の調査結果は、クエチアピンを 400 または 800 mg/日のいずれかの用量で投与した 6 週間のプラセボ対照試験に基づいています。
表 13 は、セロクエル (400 または 800 mg/日の用量) で治療された患者の 2% 以上で統合失調症の治療中 (最大 6 週間) に発生した副作用の発生率を最も近いパーセントに四捨五入して列挙しています。セロクエルで治療された患者の発生率は、プラセボで治療された患者の少なくとも 2 倍でした。
400 mg 群と比較して 800 mg 群で用量に関連する可能性があり、頻度が高かった有害反応には、めまい (8% 対 15%)、口渇 (4% 対 10%)、および頻脈 (6% 対 10%) が含まれていました。 . 11%)。
バイポーラ I マニア (10 ~ 17 歳の子供と青年)
以下の調査結果は、クエチアピンを 400 または 600 mg/日のいずれかの用量で投与した 3 週間のプラセボ対照試験に基づいています。
一般的に観察される有害反応
双極性躁病治療(最長3週間)において、小児および青年におけるクエチアピンの使用に関連して最も一般的に観察された有害反応(5%以上の発生率およびプラセボの少なくとも2倍のクエチアピン発生率)は傾眠(53%)でした。めまい(18%)、疲労(11%)、食欲増進(9%)、吐き気(8%)、嘔吐(8%)、頻脈(7%)、口渇(7%)、体重増加(6%) )。
表 14 は、セロクエル (400 または 600 mg/日の用量) で治療された患者の 2% 以上で双極性躁病の治療 (最大 3 週間) 中に発生した副作用の発生率を、最も近いパーセントに四捨五入して列挙しています。セロクエルで治療された患者の発生率は、プラセボで治療された患者の発生率よりも高かった。
400 mg 群と比較して 600 mg 群でより高い頻度で潜在的に用量に関連した有害反応には、傾眠 (50% 対 57%)、吐き気 (6% 対 10%)、および頻脈 (6% 対 10%) が含まれていました。 9%)。
錐体外路症状
思春期の統合失調症患者を対象とした短期プラセボ対照単剤療法試験(6 週間)では、錐体外路症状の総発生率は、セロクエル 100mg で 12.9%(19/147)、プラセボで 5.3%(4/75)でした。個々の有害反応(アカシジア、振戦、錐体外路障害、運動機能低下症、落ち着きのなさ、精神運動亢進、筋硬直、ジスキネジア)の発生率は、どの治療群でも4.1%を超えませんでした.小児および思春期の双極性躁病患者を対象とした短期プラセボ対照単剤療法試験(3 週間)では、錐体外路症状の合計発生率は 3.6%(7/193)またはセロクエルで 1.1%(1/90)でした。プラセボ。
表 15 は、思春期の統合失調症患者を対象とした短期プラセボ対照単剤療法試験 (6 週間) において、錐体外路症状に関連する可能性のある有害反応を示した患者のリストを示しています。
表15〜16では、ジストニアのイベントには、項部硬直、筋緊張亢進、および筋硬直が含まれていました。パーキンソニズムには、歯車の硬直と震えが含まれていました。アカシジアにはアカシジアのみが含まれます。ジスキネジア イベントには、遅発性ジスキネジア、ジスキネジア、および舞踏病アテトーシスが含まれます。その他の錐体外路イベントには、落ち着きのなさと錐体外路障害が含まれていました。
表 16 は、双極性躁病の小児および思春期の患者を対象とした短期プラセボ対照単剤療法試験 (3 週間) において、錐体外路症状に関連する有害反応を示した患者のリストを示しています。
臨床研究で観察された臨床検査、心電図、およびバイタルサインの変化
実験室の変更
好中球数
大人
クエチアピン フマル酸塩の 3368 人の患者とプラセボの 1515 人の患者を対象としたプラセボ対照単剤療法の臨床試験では、ベースラインの好中球数が正常で、少なくとも 1 つのフォローアップ検査室が利用可能な患者で、好中球数が 1.0 x 109/L 未満の発生が少なくとも 1 回発生しました。測定値は、プラセボで治療された患者の 0.1% (2/1349) と比較して、クエチアピン フマル酸塩で治療された患者で 0.3% (10/2967) でした [ 警告と注意事項 ]。
トランスアミナーゼ上昇
大人
血清トランスアミナーゼ (主に ALT) の無症候性、一過性、および可逆的な上昇が報告されています。成人を対象とした統合失調症の試験では、3~6 週間のプラセボ対照試験のプールにおいて、トランスアミナーゼ上昇が正常基準範囲の上限の 3 倍を超える患者の割合は、セロクエルで約 6% (29/483) でした。プラセボの 1% (3/194) と比較して。成人を対象とした急性双極性躁病試験では、3 週間から 12 週間のプラセボ対照試験のプールにおいて、トランスアミナーゼ上昇が正常基準範囲の上限の 3 倍を超える患者の割合は、両方のセロクエルで約 1% でした (3 /560) およびプラセボ (3/294)。これらの肝酵素の上昇は、通常、薬物治療の最初の 3 週間以内に発生し、セロクエルによる継続的な治療により、試験前のレベルに速やかに戻りました。双極性うつ病の試験では、2 つの 8 週間のプラセボ対照試験でトランスアミナーゼ上昇が正常基準範囲の上限の 3 倍を超える患者の割合は、セロクエル 50 mg で 1% (5/698)、2% (6/ 347) プラセボの場合。
ヘモグロビンの減少
大人
短期間のプラセボ対照試験では、ヘモグロビンが男性で 13 g/dL 以下、女性で 12 g/dL 以下に少なくとも 1 回減少したのは、クエチアピン治療を受けた患者の 8.3% (594/7155) で、6.2% (プラセボで治療された患者の 219/3536)。対照および非対照臨床試験のデータベースでは、クエチアピン治療を受けた患者の 11% (2277/20729) で、少なくとも 1 回、ヘモグロビンが男性で 13 g/dL 以下、女性で 12 g/dL 以下に減少しました。
尿中薬物スクリーニングへの干渉
クエチアピンを服用した患者のメタドンおよび三環系抗うつ薬の尿酵素イムノアッセイで偽陽性の結果が得られたことを示唆する文献報告があります。これらの薬物の陽性尿薬物スクリーニング結果の解釈には注意を払う必要があり、代替分析技術 (例えば、クロマトグラフィー法) による確認を考慮する必要があります。
心電図の変化
大人
プールされたプラセボ対照試験の群間比較では、QT、QTc、および PR 間隔を含む心電図パラメータの潜在的に重要な変化を経験した患者の割合において、統計的に有意なセロクエルとプラセボの差は明らかになりませんでした。しかし、頻脈の基準を満たす患者の割合は、統合失調症の治療のための 3 ~ 6 週間の 4 つのプラセボ対照臨床試験で比較され、セロクエル 100 mg の発生率が 0.6% (1 /156) プラセボの発生率。急性(単剤療法)双極性躁病試験では、頻脈の基準を満たす患者の割合は、プラセボの発生率が 0%(0/178)であるのに対し、セロクエル 200mg では 0.5%(1/192)でした。急性双極性躁病(補助)試験では、同じ基準を満たす患者の割合は、セロクエル 100mg で 0.6% (1/166)、プラセボでは 0% (0/171) でした。双極性うつ病の試験では、心拍数が毎分 120 回を超える患者はいませんでした。セロクエル 50mg の使用は、ECG によって評価された心拍数の平均増加と関連があり、プラセボ患者では 1 分あたり平均 1 回の増加と比較されました。成人におけるこのわずかな頻脈の傾向は、セロクエルが起立性変化を誘発する可能性に関連している可能性があります [参照 警告と注意事項 ]。
子供と青年
青少年を対象とした統合失調症の急性 (6 週間) 試験では、セロクエル 400 mg を投与された患者の 5.2% (3/73) およびセロクエル 800 を投与された患者の 8.5% (5/74) で心拍数の増加 (>110 bpm) が発生しました。プラセボを受けた患者の 0% (0/75) と比較して mg。心拍数の平均増加は,セロクエル 400 mg 群と 800 mg 群でそれぞれ 3.8 bpm と 11.2 bpm であったのに対し,プラセボ群では 3.3 bpm 減少した [参照 警告と注意事項 ]。
小児および青年を対象とした急性 (3 週間) の双極性躁病試験では、セロクエル 400 mg を投与された患者の 1.1% (1/89) および 4.7% (4/85) の患者で心拍数の増加 (>110 bpm) が発生しました。セロクエル 600 mg を投与された患者は、プラセボを投与された患者の 0% (0/98) と比較されました。心拍数の平均増加は,セロクエル 400 mg 群と 600 mg 群でそれぞれ 12.8 bpm と 13.4 bpm であったのに対し,プラセボ群では 1.7 bpm 減少した [参照 警告と注意事項 ]。
双極性うつ病の小児および青年(10~17 歳)を対象とした急性(8 週間)のセロクエル XR 試験では、有効性が確立されていませんが、心拍数が増加します(>110 bpm 10~12 歳および 13~17 歳)。年) は、セロクエル XR を投与された患者の 0%、プラセボを投与された患者の 1.2% で発生しました。心拍数の平均増加は、プラセボ群の 0.3 bpm と比較して、セロクエル XR では 3.4 bpm でした [ 警告と注意事項 ]。
市販後の経験
セロクエルの承認後、以下の副作用が確認されました。これらの反応は不確かな規模の集団から自発的に報告されるため、その頻度を確実に推定したり、薬物曝露との因果関係を確立したりすることは常に可能ではありません.
市場導入以降に報告された、一時的にクエチアピン療法に関連した副作用には、アナフィラキシー反応、心筋症、好酸球増多および全身症状を伴う薬物反応 (DRESS)、低ナトリウム血症、心筋炎、夜尿症、膵炎、逆行性健忘症、横紋筋融解症、抗利尿ホルモン不適合分泌症候群などがあります。 (SIADH)、スティーブンス・ジョンソン症候群(SJS)、中毒性表皮壊死融解症(TEN)、血小板数減少、重篤な肝反応(肝炎、肝壊死、肝不全を含む)、無顆粒球症、腸閉塞、イレウス、結腸虚血、尿閉、睡眠時無呼吸、および急性汎発性発疹性膿疱症(AGEP)。
薬物相互作用
クエチアピンに対する他の薬剤の効果
セロクエルを他の薬剤と組み合わせて使用するリスクは、体系的な研究で広く評価されていません。セロクエルの主な中枢神経系への影響を考えると、セロクエルを他の中枢作用薬と併用する場合は注意が必要です。セロクエル 300mg は、選択された精神病性障害の被験者を対象とした臨床試験で、アルコールの認知および運動への影響を増強したため、クエチアピンを服用している間はアルコール飲料を制限する必要があります。
クエチアピンへの暴露は、プロトタイプの CYP3A4 阻害剤 (例、ケトコナゾール、イトラコナゾール、インジナビル、リトナビル、ネファゾドンなど) によって増加し、プロトタイプの CYP3A4 誘導剤 (例、フェニトイン、カルバマゼピン、リファンピン、アバシミベ、セントジョーンズワートなど) によって減少します。 .クエチアピンが強力な CYP3A4 誘導剤または阻害剤と同時投与される場合、クエチアピンの用量調整が必要になります。
CYP3A4阻害剤
シトクロム CYP3A4 の強力な阻害剤であるケトコナゾールの同時投与により、クエチアピン曝露が大幅に増加しました。セロクエル 200mg の用量は、強力な CYP3A4 阻害剤と併用する場合、元の用量の 6 分の 1 に減量する必要があります。 投薬と管理 と 臨床薬理学 ]。
CYP3A4 インデューサー
クエチアピンと CYP3A4 インデューサーであるフェニトインの同時投与により、クエチアピンの平均経口クリアランスが 5 倍増加しました。クエチアピンとフェニトイン、または他の既知の強力な CYP3A4 誘導剤を投与されている患者では、統合失調症の症状のコントロールを維持するために、セロクエル 50mg の用量を最大 5 倍に増やす必要がある場合があります。 投薬と管理 と 臨床薬理学 ]。 CYP3A4 誘導剤が中止された場合、セロクエル 25mg の用量は 7~14 日以内に元のレベルまで減量する必要があります [ 投薬と管理 ]。
クエチアピンの薬物動態に対するいくつかの併用薬の潜在的な効果が研究された[ 臨床薬理学 ]。
他の薬剤に対するクエチアピンの効果
低血圧を誘発する可能性があるため、セロクエル 50mg は特定の降圧剤の効果を高める可能性があります。
セロクエル 25mg は、レボドパおよびドーパミン作動薬の作用に拮抗する可能性があります。
CYP 経路に基づく他の薬剤に対するセロクエルの臨床的に関連する薬物動態学的相互作用はありません。セロクエル 50mg とその代謝物は、主要な代謝 CYP (1A2、2C9、2C19、2D6、および 3A4) の非阻害剤です。
薬物乱用と依存
規制物質
セロクエルは規制物質ではありません。
乱用
セロクエルは、乱用、耐性、または身体的依存の可能性について、動物またはヒトで体系的に研究されていません.臨床試験では薬物探索行動の傾向は明らかになりませんでしたが、これらの観察結果は系統だったものではなく、この限られた経験に基づいて、CNS 活性薬物がどの程度誤用され、流用されるかを予測することはできません。および/または販売後に悪用される。したがって,薬物乱用の病歴について患者を注意深く評価し,セロクエルの誤用または乱用の徴候(耐性の発現,用量の増加,薬物探索行動など)がないか注意深く観察する必要があります。
警告
の一部として含まれています "予防" セクション
予防
認知症関連精神病の高齢患者における死亡率の増加
抗精神病薬で治療されている認知症関連精神病の高齢患者は、死亡リスクが高くなります。主に非定型抗精神病薬を服用している患者を対象とした 17 のプラセボ対照試験 (モーダル期間 10 週間) の分析により、薬物治療を受けた患者の死亡リスクは、プラセボ治療を受けた患者の死亡リスクの 1.6 ~ 1.7 倍であることが明らかになりました。典型的な 10 週間の対照試験の過程で、薬物治療を受けた患者の死亡率は約 4.5% であり、プラセボ群では約 2.6% でした。死因はさまざまであるが、ほとんどの死因は心血管疾患(例、心不全、突然死)または感染症(例、肺炎)のいずれかであると思われた。観察研究は、非定型抗精神病薬と同様に、従来の抗精神病薬による治療が死亡率を増加させる可能性があることを示唆しています。観察研究における死亡率の増加の所見が、患者のいくつかの特徴とは対照的に、抗精神病薬に起因する可能性がある程度は明らかではありません.セロクエルは、認知症関連の精神病患者の治療には承認されていません [参照 囲み警告 ]。
青少年と若年成人の自殺念慮と自殺行動
成人および小児の大うつ病性障害(MDD)の患者は、抗うつ薬を服用しているかどうかにかかわらず、うつ病の悪化および/または自殺念慮および自殺行動(自殺傾向)の出現、または行動の異常な変化を経験する可能性があります。リスクは、有意な寛解が起こるまで持続する可能性があります。自殺は、うつ病やその他の特定の精神障害の既知のリスクであり、これらの障害自体が自殺の最も強力な予測因子です。しかし、抗うつ薬が治療の初期段階で特定の患者のうつ病の悪化と自殺傾向の出現を誘発する役割を果たしている可能性があるという長年の懸念がありました.抗うつ薬 (SSRI など) の短期プラセボ対照試験のプール分析では、これらの薬が大うつ病の子供、青年、若年成人 (18 ~ 24 歳) の自殺念慮および自殺行動 (自殺傾向) のリスクを高めることが示されました。障害(MDD)およびその他の精神障害。短期間の研究では、24 歳を超える成人のプラセボと比較して、抗うつ薬による自殺のリスクの増加は示されませんでした。 65 歳以上の成人では、プラセボと比較して抗うつ薬が減少しました。
MDD、強迫性障害(OCD)、またはその他の精神障害を有する小児および青年を対象としたプラセボ対照試験のプール分析には、4,400 人を超える患者を対象とした 9 種類の抗うつ薬の合計 24 の短期試験が含まれていました。 MDD またはその他の精神障害を有する成人を対象としたプラセボ対照試験のプール分析には、77,000 人を超える患者を対象とした 11 種類の抗うつ薬の合計 295 件の短期試験 (期間の中央値は 2 か月) が含まれていました。薬物間で自殺のリスクにかなりのばらつきがありましたが、研究されたほとんどすべての薬物で若い患者が増加する傾向がありました。さまざまな適応症で自殺の絶対リスクに違いがあり、MDD での発生率が最も高かった。ただし、リスクの違い (薬物とプラセボ) は、年齢層内および適応症全体で比較的安定していました。これらのリスクの違い (治療を受けた患者 1000 人あたりの自殺傾向の症例数における薬物とプラセボの違い) を表 2 に示します。
どの小児科試験でも自殺は発生していません。成人試験では自殺がありましたが、自殺に対する薬物の影響について結論を出すには十分な数ではありませんでした。
自殺リスクが長期間の使用、つまり数ヶ月以上の使用に及ぶかどうかは不明です。しかし、うつ病の成人におけるプラセボ対照維持試験から、抗うつ薬の使用がうつ病の再発を遅らせることができるという実質的な証拠があります.
適応症を問わず抗うつ薬で治療されているすべての患者は、特に薬物療法のコースの最初の数か月間、または用量変更時に、臨床的悪化、自殺傾向、行動の異常な変化について適切に監視し、注意深く観察する必要があります。または減少します。
大うつ病性障害で抗うつ薬を服用している成人および小児患者においても、不安、興奮、パニック発作、不眠症、易怒性、敵意、攻撃性、衝動性、アカシジア(精神運動の落ち着きのなさ)、軽躁病、躁病の症状が報告されています。他の適応症に関しては、精神医学的および非精神医学的の両方.そのような症状の出現と、うつ病の悪化および/または自殺衝動の出現との間の因果関係は確立されていませんが、そのような症状が自殺傾向の出現の前兆を表している可能性があるという懸念があります.
うつ病が持続的に悪化している患者、またはうつ病や自殺傾向の悪化の前兆である可能性のある緊急の自殺願望または症状を経験している患者では、場合によっては投薬を中止することを含め、治療計画の変更を検討する必要があります。特にこれらの症状が重度で突然の場合発症したか、患者の症状の一部ではありませんでした。
大うつ病性障害またはその他の適応症(精神科および非精神科の両方)で抗うつ薬による治療を受けている患者の家族および介護者は、動揺、易怒性、行動の異常な変化、およびその他の症状の出現について患者を監視する必要性について注意を喚起する必要があります。自殺傾向の出現と同様に、そのような症状を直ちに医療提供者に報告すること。このようなモニタリングには、家族や介護者による毎日の観察が含まれる必要があります。 セロクエルの処方は、過剰摂取のリスクを軽減するために、適切な患者管理と一致する最小量の錠剤用に作成する必要があります。
双極性障害の患者のスクリーニング
大うつ病エピソードは、双極性障害の最初の症状である可能性があります。このようなエピソードを抗うつ薬のみで治療すると、双極性障害のリスクがある患者に混合/躁病エピソードが発生する可能性が高くなる可能性があると一般的に考えられています (対照試験では確立されていません)。上記の症状のいずれかがそのような変換を表しているかどうかは不明です。ただし、セロクエルを含む抗うつ薬による治療を開始する前に、抑うつ症状のある患者を適切にスクリーニングして、双極性障害のリスクがあるかどうかを判断する必要があります。そのようなスクリーニングには、自殺、双極性障害、うつ病の家族歴など、詳細な精神病歴を含める必要があります。
認知症関連精神病の高齢患者における脳卒中を含む脳血管有害反応
認知症の高齢者を対象としたリスペリドン、アリピプラゾール、およびオランザピンによるプラセボ対照試験では、プラセボ治療を受けた被験者と比較して、致死率を含む脳血管有害反応(脳血管障害および一過性脳虚血発作)の発生率が高かった.セロクエルは認知症関連の精神病患者の治療には承認されていない [参照 囲み警告 と 認知症関連精神病の高齢患者における死亡率の増加 ]。
悪性症候群(NMS)
セロクエルを含む抗精神病薬の投与に関連して、神経弛緩性悪性症候群 (NMS) と呼ばれることもある致命的な複合症状が報告されています。セロクエルでは、まれに NMS の症例が報告されています。 NMS の臨床症状は、高熱、筋肉の硬直、精神状態の変化、および自律神経系の不安定性 (不規則な脈拍または血圧、頻脈、発汗、および不整脈) の証拠です。追加の徴候には、クレアチニンホスホキナーゼの上昇、ミオグロビン尿症 (横紋筋融解症)、および急性腎不全が含まれる場合があります。
この症候群の患者の診断評価は複雑です。診断を下す際には、臨床症状に深刻な医学的疾患(例、肺炎、全身感染症など)と未治療または不十分な治療を受けた錐体外路徴候および症状(EPS)の両方が含まれる症例を除外することが重要です。鑑別診断におけるその他の重要な考慮事項には、中枢性抗コリン作動性毒性、熱射病、薬物熱、および原発性中枢神経系 (CNS) の病理が含まれます。
NMS の管理には次のことを含めるべきです。 2) 集中的な対症療法と医学的モニタリング。 3) 特定の治療法が利用可能な付随する深刻な医学的問題の治療。 NMS の特定の薬理学的治療レジメンについては、一般的な合意はありません。
患者が NMS からの回復後に抗精神病薬治療を必要とする場合、薬物療法の再導入の可能性を慎重に検討する必要があります。 NMSの再発が報告されているため、患者を注意深く監視する必要があります。
代謝変化
非定型抗精神病薬は、高血糖/真性糖尿病、脂質異常症、および体重増加を含む代謝変化と関連しています。このクラスのすべての薬が何らかの代謝変化を引き起こすことが示されていますが、各薬には独自のリスクプロファイルがあります.一部の患者では、体重、血糖、および脂質の複数の代謝パラメーターの悪化が臨床研究で観察されました。これらの代謝プロファイルの変化は、臨床的に適切に管理する必要があります。
高血糖と糖尿病
クエチアピンを含む非定型抗精神病薬で治療された患者では、ケトアシドーシスまたは高浸透圧性昏睡または死亡を伴う場合もある極度の高血糖が報告されています。非定型抗精神病薬の使用とグルコース異常との関係の評価は、統合失調症患者における真性糖尿病のバックグラウンドリスクの増加の可能性と、一般集団における真性糖尿病の発生率の増加の可能性によって複雑になっています。これらの交絡因子を考慮すると、非定型抗精神病薬の使用と高血糖関連の有害反応との関係は完全には理解されていません。しかし、疫学的研究は、非定型抗精神病薬で治療された患者における高血糖関連の有害反応のリスクの増加を示唆しています。非定型抗精神病薬で治療された患者における高血糖関連の有害反応の正確なリスク推定値は入手できません。
非定型抗精神病薬を開始した真性糖尿病の診断が確立された患者は、定期的に血糖コントロールの悪化を監視する必要があります。糖尿病の危険因子(例、肥満、糖尿病の家族歴)を持ち、非定型抗精神病薬による治療を開始する患者は、治療開始時と治療中定期的に空腹時血糖検査を受けるべきです。非定型抗精神病薬で治療された患者は、多飲、多尿、多食、衰弱などの高血糖の症状を監視する必要があります。非定型抗精神病薬による治療中に高血糖の症状を発症した患者は、空腹時血糖検査を受ける必要があります。場合によっては、非定型抗精神病薬を中止すると高血糖が解消されます。しかし、一部の患者は、疑わしい薬物の中止にもかかわらず、抗糖尿病治療の継続を必要としました。
大人
すべての患者の経口耐糖能試験で血糖状態を評価するように設計された 24 週間の試験 (実薬対照、セロクエルで治療された 115 人の患者) では、24 週目にグルコースチャレンジ後の血糖値が 200 mg/dL 以上の発生率でした。 1.7%、空腹時血糖値≧126mg/dLの発生率は2.6%でした。ベースラインからの空腹時血糖の平均変化は 3.2 mg/dL であり、クエチアピンのベースラインからの 2 時間血糖の平均変化は -1.8 mg/dL でした。
双極 I 型障害の維持を目的とした 2 つの長期プラセボ対照無作為化離脱臨床試験では、セロクエル (646 人の患者) で平均 213 日間、プラセボ (680 人の患者) で 152 日間、ベースラインからのグルコースの平均変化は +5.0 mg でした。セロクエル 100mg では /dL、プラセボでは –0.05 mg/dL。食事から 8 時間以上経過した患者の血糖値上昇 (≥126 mg/dL) の曝露調整率 (ただし、一部の患者は絶食期間中の水分からのカロリー摂取を排除できなかった可能性があります) は、100 あたり 18.0 でした。 100 患者年あたりのセロクエルの患者年数 (患者の 10.7%; n=556) およびプラセボの患者年数は 9.5 (患者の 4.6%; n=581)。
子供と青年
統合失調症の思春期患者(13~17 歳)(6 週間)を対象としたプラセボ対照セロクエル 25mg 単剤療法試験では、プラセボ(n=67)と比較したセロクエル(n=138)の空腹時血糖値の平均変化–0.75 mg/dL 対 –1.70 mg/dL でした。双極性躁病の小児および青年患者 (10~17 歳) を対象としたプラセボ対照セロクエル 300mg 単剤療法試験 (3 週間) では、セロクエル (n=170) の空腹時血糖値の平均変化は、プラセボ (n =81) は、-1.17 mg/dL に対して 3.62 mg/dL でした。ベースライン正常空腹時血糖値 (
有効性が確立されていない双極性うつ病の小児および青年患者(10~17 歳)を対象としたセロクエル XR 単剤療法のプラセボ対照試験(8 週間)では、セロクエル 300mg XR の空腹時血糖値の平均変化(n= 60) は、プラセボ (n=62) と比較して、1.6 mg/dL に対して 1.8 mg/dL でした。この研究では、セロクエル XR またはプラセボ投与群に、ベースラインの空腹時血糖値が正常 (100 mg/dL および
脂質異常症
大人
表 4 は、セロクエルの臨床試験において、総コレステロール、トリグリセリド、LDL-コレステロール、および HDL-コレステロールがベースラインから変化した成人患者の割合を示しています。
子供と青年
表 5 は、セロクエルの臨床試験において、総コレステロール、トリグリセリド、LDL コレステロール、および HDL コレステロールがベースラインから変化した小児および青年の割合を示しています。
双極性うつ病の小児および青年患者(10~17 歳)を対象としたプラセボ対照セロクエル XR 単剤療法試験(8 週間)では、有効性が確立されていませんでしたが、総コレステロール値が変化した小児および青年の割合(≥200 mg/dL)、トリグリセリド (≥150 mg/dL)、LDL-コレステロール (≥ 130 mg/dL)、および HDL-コレステロール (≤40 mg/dL) をベースラインから臨床的に有意なレベルまで: 総コレステロール 8% (7 /83) セロクエル 50mg XR 対プラセボの 6% (5/84);トリグリセリド 28% (22/80) セロクエル 100mg XR vs. 9% (7/82) プラセボ;セロクエル XR の LDL コレステロール 2% (2/86) 対プラセボの 4% (3/85) およびセロクエル XR の HDL コレステロール 20% (13/65) 対プラセボの 15% (11/74)。
体重の増加
臨床試験では、体重の増加が観察されています。クエチアピンを投与されている患者は、定期的に体重をモニタリングする必要があります。
大人
セロクエル 50mg の臨床試験では、以下のような体重増加が報告されています。
子供と青年
セロクエル 25mg を用いた 2 つの臨床試験 (双極性躁病患者と統合失調症患者) で報告された体重増加を表 7 に示します。
統合失調症試験における体重の平均変化は、セロクエル 25 mg 群で 2.0 kg、プラセボ群で -0.4 kg、双極性躁病試験では、セロクエル 200 mg 群で 1.7 kg、プラセボ群で 0.4 kg でした。
上記の 2 つの小児科試験から患者を登録した非盲検試験では、患者の 63% (241/380) がセロクエルによる 26 週間の治療を完了しました。 26 週間の治療後、体重の平均増加は 4.4 kg でした。患者の 45% が体重の 7% 以上増加しましたが、通常の成長に合わせて調整されていません。 26 週間にわたる正常な成長に合わせて調整するために、BMI のベースラインからの少なくとも 0.5 標準偏差の増加が、臨床的に有意な変化の尺度として使用されました。セロクエルの患者の 18.3% が、26 週間の治療後にこの基準を満たしました。
有効性が確立されていない双極性うつ病の小児および青年(10~17 歳)を対象としたセロクエル XR の臨床試験では、体重が常に体重の 7% 以上増加した患者の割合は 15%(セロクエル 25mg XR では 14/92)、プラセボでは 10% (10/100)。体重の平均変化は、セロクエル 50mg XR 群で 1.4 kg であったのに対し、プラセボ群では 0.6 kg でした。
小児患者にセロクエル 50mg を適応症として治療する場合、正常な成長に期待される体重増加と比較して体重増加を評価する必要があります。
遅発性ジスキネジア
クエチアピンを含む抗精神病薬で治療された患者では、潜在的に不可逆的で不随意の運動障害症候群が発生する可能性があります。この症候群の有病率は高齢者、特に年配の女性の間で最も高いようですが、抗精神病薬治療の開始時に、どの患者がこの症候群を発症する可能性があるかを予測するために有病率の推定に頼ることは不可能です。抗精神病薬製品が遅発性ジスキネジアを引き起こす可能性が異なるかどうかは不明です。
遅発性ジスキネジアを発症するリスクとそれが不可逆的になる可能性は、治療期間と患者に投与される抗精神病薬の総累積用量が増加するにつれて増加すると考えられています。しかし、この症候群は、低用量での比較的短い治療期間の後に発生する可能性があり、あまり一般的ではありませんが、治療の中止後に発生することさえあります.
遅発性ジスキネジアは、抗精神病薬の治療を中止すると、部分的または完全に寛解することがあります。しかし、抗精神病薬の治療自体が、症候群の徴候や症状を抑制 (または部分的に抑制) する可能性があり、それによって潜在的なプロセスを覆い隠す可能性があります。症状の抑制が症候群の長期経過に及ぼす影響は不明です。
これらの考慮事項を考慮すると、遅発性ジスキネジアの発生を最小限に抑える可能性が最も高い方法でセロクエル 50mg を処方する必要があります。慢性抗精神病治療は、一般に、(1) 抗精神病薬に反応することが知られている慢性疾患に苦しんでいると思われる患者、および (2) 同等に効果的であるが潜在的に害の少ない代替治療が利用できないか適切でない患者に限定されるべきです。 .慢性的な治療を必要とする患者では、満足のいく臨床反応をもたらす最小用量と最短の治療期間を追求する必要があります。継続的な治療の必要性は、定期的に再評価する必要があります。

セロクエル 200mg を服用している患者に遅発性ジスキネジアの徴候と症状が現れた場合は、投薬の中止を考慮する必要があります。しかし、一部の患者は、症候群の存在にもかかわらず、セロクエル 200mg による治療を必要とする場合があります。
低血圧
クエチアピンは、おそらくそのα1-アドレナリン拮抗薬の特性を反映して、めまい、頻脈、および一部の患者では失神を伴う起立性低血圧を誘発する可能性があります。セロクエルで治療を受けた患者の 1% (28/3265) で失神が報告されたのに対し、プラセボでは 0.2% (2/954)、実薬対照薬では約 0.4% (2/527) でした。起立性低血圧、めまい、失神により転倒することがあります。
セロクエル 25 mg は、既知の心血管疾患 (心筋梗塞または虚血性心疾患の病歴、心不全、または伝導異常)、脳血管疾患、または患者が低血圧になりやすい状態 (脱水、循環血液量減少、および降圧薬)。起立性低血圧および失神のリスクは、初回用量を 1 日 2 回 25 mg に制限することによって最小限に抑えることができます。 投薬と管理 ]。目標用量への漸増中に低血圧が発生した場合は、漸増スケジュールの前の用量に戻すことが適切です。
滝
セロクエルを含む非定型抗精神病薬は、傾眠、起立性低血圧、運動および感覚の不安定性を引き起こす可能性があり、転倒につながる可能性があり、その結果、骨折やその他の怪我につながる可能性があります。これらの影響を悪化させる可能性のある疾患、状態、または薬を服用している患者については、抗精神病治療を開始するときに転倒リスク評価を完了し、長期の抗精神病治療を受けている患者については繰り返します。
血圧の上昇(小児および青年)
統合失調症 (6 週間) または双極性躁病 (3 週間) の小児および青年を対象としたプラセボ対照試験では、収縮期血圧 (≧20 mmHg) の任意の時点での上昇の発生率は 15.2% (51/335 ) セロクエル 50mg では 5.5% (9/163)、プラセボでは 5.5% (9/163)。拡張期血圧(≥10 mmHg)の任意の時点での上昇の発生率は、セロクエルで 40.6%(136/335)、プラセボで 24.5%(40/163)でした。 26 週間の非盲検臨床試験では、高血圧の病歴が報告されている 1 人の子供が高血圧の危機を経験しました。小児および青年の血圧は、治療の開始時および治療中に定期的に測定する必要があります。
双極性うつ病の小児および青年(10~17 歳)を対象としたプラセボ対照セロクエル XR 臨床試験(期間 8 週間)では、有効性が確立されていませんでしたが、収縮期血圧(≧ 20 mmHg) は、セロクエル XR で 6.5% (6/92)、プラセボで 6.0% (6/100) でした。任意の時点での拡張期血圧 (≥10 mmHg) の上昇の発生率は、セロクエル 25mg XR で 46.7% (43/92)、プラセボで 36.0% (36/100) でした。
白血球減少症、好中球減少症、および無顆粒球症
臨床試験および市販後の経験では、セロクエルを含む非定型抗精神病薬に一時的に関連する白血球減少/好中球減少のイベントが報告されています。無顆粒球症が報告されています。
無顆粒球症 (絶対好中球数
白血球減少症/好中球減少症の可能性のある危険因子には、既存の低白血球数 (WBC) および薬剤誘発性白血球減少症/好中球減少症の病歴が含まれます。既存の低 WBC または薬剤誘発性白血球減少症/好中球減少症の病歴のある患者は、治療開始から最初の数か月間、全血球計算 (CBC) を頻繁にモニタリングし、白血球減少の最初の兆候が見られたらセロクエル 50mg の投与を中止する必要があります。他の原因となる要因がないこと。
好中球減少症の患者は、発熱やその他の症状や感染の徴候について注意深く監視し、そのような症状や徴候が発生した場合は速やかに治療する必要があります。重度の好中球減少症 (絶対好中球数
白内障
白内障の発症は、慢性的なイヌの研究でクエチアピン治療に関連して観察された[参照 非臨床毒性学 ]。セロクエル 100mg の長期投与中、成人、小児、青年でも水晶体変化が観察されていますが、セロクエル 200mg の使用との因果関係は確立されていません。それにもかかわらず、現時点ではレンズの変化の可能性を排除することはできません.したがって、細隙灯検査やその他の適切に感度の高い方法など、白内障の形成を検出するのに適切な方法による水晶体の検査は、治療の開始時またはその直後、および長期治療中は 6 か月間隔で行うことをお勧めします。
QT延長
臨床試験では、クエチアピンは QT 間隔の持続的な増加とは関連していませんでした。ただし、QT 効果は徹底的な QT 研究で体系的に評価されていません。市販後の経験では、クエチアピンを過剰摂取した患者で QT 延長が報告された例があった [ 過剰摂取 ]、併存疾患のある患者、および電解質の不均衡を引き起こしたり、QT間隔を増加させることが知られている薬を服用している患者[参照 薬物相互作用 ]。
クエチアピンの使用は、クラス 1A 抗不整脈薬(例、キニジン、プロカインアミド)またはクラス III 抗不整脈薬(例、アミオダロン、ソタロール)、抗精神病薬(例、ジプラシドン、クロルプロマジン、チオリダジン)、抗生物質 (例、ガチフロキサシン、モキシフロキサシン)、または QTc 間隔を延長することが知られている他のクラスの薬物 (例、ペンタミジン、酢酸レボメタジル、メタドン)。
クエチアピンはまた、以下を含むトルサード ド ポワントおよび/または突然死の発生リスクを高める可能性のある状況では避けるべきです。 (2) 低カリウム血症または低マグネシウム血症; (3)QTc間隔を延長する他の薬剤の併用。 (4)QT間隔の先天性延長の存在。
QT延長のリスクが高い患者(例、心血管疾患、QT延長の家族歴、高齢者、うっ血性心不全、心肥大など)にクエチアピンを処方する場合も注意が必要です。
発作
臨床試験中、セロクエルで治療された患者の 0.5% (20/3490) で発作が発生したのに対し、プラセボでは 0.2% (2/954)、実薬対照薬では 0.7% (4/527) でした。他の抗精神病薬と同様に、セロクエル 25mg は、発作の既往歴のある患者、またはアルツハイマー型認知症などの発作閾値を低下させる可能性のある状態の患者には慎重に使用する必要があります。発作閾値を下げる条件は、65 歳以上の集団でより一般的である可能性があります。
甲状腺機能低下症
大人
クエチアピンの臨床試験では、甲状腺ホルモンレベルが用量依存的に減少することが実証されました。治療用量範囲の上限で約 20% の総チロキシンおよび遊離サイロキシン (T4) の減少は、治療の最初の 6 週間で最大であり、より長期的な治療中に順応または進行することなく維持されました。ほぼすべてのケースで、クエチアピン治療の中止は、治療期間に関係なく、総 T4 および遊離 T4 に対する効果の逆転と関連していました。クエチアピンが甲状腺軸に影響を与えるメカニズムは不明です。視床下部-下垂体軸に影響がある場合、TSH のみの測定では患者の甲状腺状態を正確に反映できない可能性があります。したがって、臨床評価に加えて、TSH と遊離 T4 の両方をベースライン時と追跡時に測定する必要があります。
セロクエル 50mg がリチウムまたはジバルプロエックスに追加された躁病補助研究では、セロクエル 200mg で治療された患者の 12% (24/196) が、プラセボで治療された患者の 7% (15/203) で TSH レベルが上昇した。 TSH レベルが上昇したセロクエル 300mg 治療患者のうち、3 人は同時に遊離 T4 レベルが低かった (遊離 T4
セロクエル患者の約 0.7% (26/3489) が、単剤療法の研究で TSH の上昇を経験しました。一部の TSH 患者では、必要な甲状腺置換治療が増加します。
すべてのクエチアピン試験で、甲状腺ホルモンと TSH のシフトの発生率は 1: 遊離 T4 の減少 (5mIU/L)、4.9% (956/19412)。 TBGが測定された8人の患者では、TBGのレベルは変化していませんでした。
表 8 は、短期間のプラセボ対照臨床試験におけるこれらのシフトの発生率を示しています。
短期のプラセボ対照単剤療法試験では、T3 と TSH の相互シフトの発生率は、クエチアピン (1/4800) とプラセボ (0/2190) の両方で 0.0% であり、T4 と TSH のシフトは 0.1% でした (7 /6154) 対プラセボの 0.0% (1/3007)。
子供と青年
小児および思春期の統合失調症患者(6 週間)または双極性躁病患者(3 週間)を対象とした急性プラセボ対照試験では、セロクエル 100mg 投与患者とプラセボ投与患者の任意の時点での甲状腺機能値の変化の発生率上昇した TSH は、それぞれ 2.9% (8/280) 対 0.7% (1/138) であり、総チロキシンの低下は、それぞれ 2.8% (8/289) 対 0% (0/145) でした。 TSH レベルが上昇したセロクエル治療を受けた患者のうち 1 人は、治療終了時に同時に遊離 T4 レベルが低かった。
高プロラクチン血症
大人
クエチアピンの臨床試験中、臨床的に有意な値へのプロラクチンレベルのシフトの発生率は、プラセボの 2.6% (51/1968) と比較して、クエチアピンで治療された患者の 3.6% (158/4416) で発生しました。
子供と青年
双極性躁病(3週間)または統合失調症(6週間)の小児および青年患者を対象とした急性プラセボ対照試験では、プロラクチンレベルのシフトの発生率は、ある値(> 20μg/ Lの男性; > 26μg) /L 女性の任意の時点で) は、セロクエルで 13.4% (18/134)、男性でプラセボで 4% (3/75)、セロクエル 25 mg で 8.7% (9/104)、0% (0/39) でした。女性のプラセボ用。
ドーパミン D2 受容体に拮抗する他の薬剤と同様に、セロクエル 300mg は一部の患者でプロラクチンレベルを上昇させ、その上昇は慢性投与中に持続する可能性があります。高プロラクチン血症は、病因に関係なく、視床下部の GnRH を抑制し、下垂体のゴナドトロピン分泌を低下させる可能性があります。これは、女性と男性の両方の患者で生殖腺のステロイド産生を損なうことにより、生殖機能を阻害する可能性があります。プロラクチン上昇化合物を投与されている患者では、乳汁漏出症、無月経、女性化乳房、およびインポテンスが報告されています。性腺機能低下症に関連する長期にわたる高プロラクチン血症は、女性と男性の両方の被験者で骨密度の低下につながる可能性があります。
組織培養実験では、ヒト乳癌の約 3 分の 1 が in vitro でプロラクチン依存性であることが示されています。これは、以前に乳癌が発見された患者でこれらの薬剤の処方が考慮される場合、潜在的に重要な要素です。プロラクチン放出を増加させる化合物と同様に、乳腺および膵島細胞腫瘍 (乳腺癌、脳下垂体、および膵臓腺腫) が、マウスおよびラットで実施された発がん性研究で観察されました。これまでに実施された臨床研究も疫学研究も、このクラスの薬物の慢性投与とヒトの腫瘍形成との関連を示していませんが、入手可能な証拠は限定的すぎて決定的ではありません[ 非臨床毒性学 ]。
認知障害および運動障害の可能性
傾眠は、セロクエルで治療された患者で報告された一般的な有害事象であり、特に初回用量漸増の 3~5 日間に報告されました。統合失調症の試験では、プラセボ患者の 11% (22/206) と比較して、セロクエル 200mg の患者の 18% (89/510) で傾眠が報告されました。セロクエル 200mg を単剤療法として使用した急性双極性躁病試験では、プラセボ患者の 4% と比較して、セロクエル 300mg の患者の 16% (34/209) で傾眠が報告されました。補助療法としてセロクエルを使用した急性双極性躁病試験では、プラセボ患者の 9% (19/203) と比較して、セロクエルの患者の 34% (66/196) で傾眠が報告されました。双極うつ病試験では、プラセボ患者の 15% (51/347) と比較して、セロクエル 50mg の患者の 57% (398/698) で傾眠が報告されました。セロクエル 25mg は判断力,思考力,または運動能力を損なう可能性があるため,患者は,セロクエル療法が有効であると合理的に確信できるまでは,自動車(自動車を含む)の運転や危険な機械の操作などの精神的覚醒を必要とする活動を行うことについて注意する必要があります。それらに悪影響を与えません。傾眠は転倒につながる可能性があります。
体温調節
セロクエル 100mg については報告されていませんが、深部体温を下げる体の能力の混乱は、抗精神病薬に起因しています。セロクエル 300mg を処方する際は、激しい運動、極度の暑さへの曝露、抗コリン作用を伴う併用薬の服用、脱水症状など、深部体温の上昇に寄与する可能性のある状態を経験する可能性のある患者に適切な注意を払うことをお勧めします。
嚥下障害
食道の運動障害と誤嚥は、抗精神病薬の使用に関連付けられています。誤嚥性肺炎は、高齢患者、特に進行したアルツハイマー型認知症患者の罹患率と死亡率の一般的な原因です。セロクエル 100mg およびその他の抗精神病薬は、誤嚥性肺炎のリスクがある患者には慎重に使用する必要があります。
中断症候群
セロクエルを含む非定型抗精神病薬の突然の中止後に、不眠症、吐き気、嘔吐などの急性離脱症状が報告されています。セロクエル 25mg XR による短期間のプラセボ対照単剤療法臨床試験では、中止症状を評価する中止フェーズが含まれ、突然の中止後に 1 つ以上の中止症状を経験した患者の総発生率は、セロクエル XR で 12.1% (241/1993) でした。プラセボでは 6.7% (71/1065)。個々の有害反応(不眠症、吐き気、頭痛、下痢、嘔吐、めまい、過敏症)の発生率は、どの治療群でも 5.3% を超えず、通常は中止後 1 週間で解消しました。徐々に撤退することをお勧めします。 [見る 特定の集団での使用 ]
抗コリン作用(抗ムスカリン作用)
クエチアピンの活性代謝物であるノルクエチアピンは、いくつかのムスカリン受容体サブタイプに対して中程度から強い親和性を持っています。これは、セロクエル 25mg を治療用量で使用したり、他の抗コリン薬と併用したり、過剰摂取したりすると、抗コリン薬の副作用につながります。セロクエルは、抗コリン作用(抗ムスカリン作用)のある薬を服用している患者には注意して使用する必要があります[参照 過剰摂取 と 臨床薬理学 ]。
便秘は、クエチアピンで治療された患者で一般的に報告された有害事象であり、腸閉塞の危険因子を表しています.腸閉塞はクエチアピンで報告されており、腸の運動性を低下させる複数の併用薬を受けていた患者の致命的な報告が含まれています.
セロクエル 300mg は、尿閉、臨床的に重大な前立腺肥大症、便秘、または眼圧上昇の現在の診断または既往歴のある患者には注意して使用する必要があります。
患者相談情報
患者に FDA 承認の患者ラベル (投薬ガイド) を読むようにアドバイスします。
セロクエルの服用中に次の問題が発生した場合は、患者に注意を促し、処方者に警告するように依頼してください。
認知症関連精神病の高齢患者における死亡率の増加 非定型抗精神病薬で治療されている認知症関連精神病の高齢患者は、プラセボと比較して死亡リスクが高いことを患者と介護者に通知する必要があります。クエチアピンは、認知症関連精神病の高齢患者には承認されていない[参照 警告と注意事項 ]。
自殺念慮と行動
患者、その家族、および介護者は、不安、動揺、パニック発作、不眠症、過敏性、敵意、攻撃性、衝動性、アカシジア(精神運動の落ち着きのなさ)、軽躁病、躁病、その他の行動の異常な変化の出現に注意するよう奨励されるべきである. 、うつ病の悪化、および自殺念慮、特に抗うつ薬治療中の初期および用量が上下に調整された場合。患者の家族や介護者は、変化が突然起こる可能性があるため、日常的にそのような症状の出現を探すようにアドバイスされるべきです.このような症状は、患者の処方者または医療専門家に報告する必要があります。特に、症状が深刻である場合、突然発症する場合、または患者の症状の一部ではない場合はそうです。これらのような症状は、自殺念慮や行動のリスクの増加と関連している可能性があり、非常に綿密な監視と、場合によっては投薬の変更が必要であることを示しています[参照 警告と注意事項 ]。
悪性症候群(NMS)
患者は、NMS に関連している可能性のある徴候や症状を医師に報告するようにアドバイスされるべきです。これらには、筋肉のこわばりや高熱が含まれる場合があります[ 警告と注意事項 ]。
高血糖と糖尿病
患者は、高血糖(高血糖)および真性糖尿病の症状に注意する必要があります。糖尿病と診断された患者、糖尿病の危険因子を有する患者、または治療中にこれらの症状を発症した患者は、治療の開始時および治療中に定期的に血糖値を監視する必要があります[参照 警告と注意事項 ]。
高脂血症
患者には、総コレステロール、LDL コレステロール、およびトリグリセリドの上昇と、HDL コレステロールの低下が起こる可能性があることを説明する必要があります。患者は、治療の開始時および治療中に定期的に脂質プロファイルをモニタリングする必要があります[参照 警告と注意事項 ]。
体重の増加
体重増加を経験する可能性があることを患者に説明する必要があります。患者は定期的に体重を測定する必要があります[参照 警告と注意事項 ]。
起立性低血圧
起立性低血圧(症状には、立ちくらみや立ちくらみが含まれ、転倒につながる可能性があります)、特に最初の用量漸増期間中、および治療の再開時または用量の増加時に、患者に注意する必要があります[見る 警告と注意事項 ]。
小児および青年における血圧上昇
小児および思春期の患者は、治療の開始時および治療中に定期的に血圧を測定する必要があります[参照 警告と注意事項 ]。
白血球減少症/好中球減少症
既存の低 WBC または薬剤性白血球減少症 / 好中球減少症の病歴がある患者には、セロクエルを服用している間は CBC を監視するようにアドバイスする必要があります。発熱、インフルエンザのような症状、喉の痛み、またはその他の感染症がある場合は、WBC が非常に低いことが原因である可能性があるため、できるだけ早く医師に相談するよう患者にアドバイスする必要があります。中止された、および/または与えられる治療[を参照してください 警告と注意事項 ]。
認知機能および運動機能への干渉
患者は、特に最初の用量漸増期間中に、傾眠または鎮静(転倒につながる可能性がある)のリスクについて知らされるべきである.患者は、クエチアピン療法が患者に悪影響を及ぼさないことを合理的に確信できるまで、自動車(自動車を含む)の運転や機械の操作など、精神的覚醒を必要とする活動を行うことについて注意する必要があります。 警告と注意事項 ]。
熱暴露と脱水
過熱や脱水を避けるための適切なケアについて、患者に助言する必要があります。 警告と注意事項 ]。
併用薬
他の医薬品と同様に、処方薬または市販薬を服用している、または服用する予定がある場合は、医師に通知するよう患者に助言する必要があります。 薬物相互作用 ]。
妊娠
妊娠中の女性がセロクエルの治療中に妊娠した場合、または妊娠する予定がある場合は、医療提供者に通知するようにアドバイスしてください。セロクエルが新生児に錐体外路症状および/または禁断症状 (激越、筋緊張亢進、筋緊張低下、振戦、傾眠、呼吸困難、および摂食障害) を引き起こす可能性があることを患者に助言する。妊娠中にセロクエル 25 mg に曝露した女性の妊娠転帰を監視する妊娠登録簿があることを患者に助言する [ 特定の集団での使用 ]。
不妊
セロクエルが血清プロラクチンレベルの上昇により生殖能力を損なう可能性があることを女性に助言する.生殖能力への影響は可逆的です [ 特定の集団での使用 ]。
包括的な治療プログラムの必要性
セロクエルは、統合失調症および小児双極性障害の青少年のための総合治療プログラムの不可欠な部分として適応されており、他の対策 (心理学的、教育的、および社会的) を含む場合があります。セロクエル 100mg の有効性と安全性は、統合失調症の 13 歳未満の小児患者または双極性躁病の 10 歳未満の小児患者では確立されていません。適切な教育配置が不可欠であり、心理社会的介入が役立つことがよくあります。非定型抗精神病薬を処方する決定は、患者の症状の慢性性と重症度に関する医師の評価に依存します[参照 適応症 ]。
非臨床毒性学
発がん、突然変異誘発、生殖能力の障害
発がん
発がん性試験は、C57BL マウスおよび Wistar ラットで実施されました。クエチアピンは、マウスには 20、75、250、および 750 mg/kg の用量で、ラットには 25、75、および 250 mg/kg の用量で 2 年間強制経口投与されました。これらの用量は、mg/m2 体表面積 (マウス) に基づく 800 mg/日の MRHD の 0.1、0.5、1.5、および 4.5 倍、または mg/m2 体表面積に基づく MRHD の 0.3、1、および 3 倍に相当します。 (ネズミ)。 mg/m2 体表面積に基づいて MRHD の 1.5 倍および 4.5 倍の用量で雄マウスに、mg/m2 体表面積に基づいて MRHD の 3 倍の用量で雄ラットに甲状腺濾胞腺腫の統計的に有意な増加があった。乳腺腺癌は、試験したすべての用量で雌ラットで統計的に有意に増加した (mg/m2 体表面積に基づく MRHD の 0.3、1、および 3 倍)。
甲状腺濾胞細胞腺腫は、げっ歯類の肝臓によるサイロキシンの代謝とクリアランスの亢進に起因する甲状腺刺激ホルモン (TSH) による甲状腺の慢性的な刺激が原因である可能性があります。この機序と一致する TSH、チロキシン、およびチロキシンクリアランスの変化が、ラットおよびマウスの亜慢性毒性試験とラットの 1 年間毒性試験で観察された。ただし、これらの研究の結果は決定的なものではありませんでした。甲状腺濾胞細胞腺腫の増加とヒトのリスクとの関連性は、どのような機序によるものであっても不明です。
抗精神病薬は、げっ歯類のプロラクチンレベルを慢性的に上昇させることが示されています。 1 年間の毒性試験における血清測定では、クエチアピンが血清プロラクチン濃度の中央値を雄ラットで最大 32 倍、雌ラットで最大 13 倍増加させたことが示されました。乳腺腫瘍の増加は、他の抗精神病薬の慢性投与後のげっ歯類で発見されており、プロラクチン媒介と考えられています。このラットにおけるプロラクチン介在性乳腺腫瘍の発生率の増加とヒトのリスクとの関連性は不明である[参照 警告と注意事項 ]。
突然変異誘発
クエチアピンは、標準的な遺伝毒性試験で変異原性または染色体異常誘発性ではありませんでした。クエチアピンの変異原性の可能性は、in vitro Ames 細菌遺伝子変異アッセイおよび in vitro 哺乳動物遺伝子変異アッセイでチャイニーズハムスター卵巣細胞で試験されました。クエチアピンの染色体異常誘発能は、培養ヒトリンパ球の in vitro 染色体異常試験およびラットの in vivo 骨髄小核試験で最大 500 mg/kg で試験されました。これは、mg/m2 に基づく最大推奨ヒト用量の 6 倍です。体表面積。
生殖能力の障害
クエチアピンは、オスの Sprague-Dawley ラットに 50 および 150 mg/kg の経口投与で交配および受胎能を低下させ、mg/m2 体表面積に基づいて 800 mg/日の MRHD の約 1 倍および 3 倍でした。薬物関連の影響には、交尾までの間隔の増加、および受精の成功に必要な交配回数の増加が含まれていました。これらの効果は、治療なしで 2 週間後でも MRHD の 3 倍で観察され続けました。オスのラットにおける交尾障害および受胎能の障害に対する無影響用量は、25 mg/kg、または mg/m2 体表面積に基づく MRHD 用量の 0.3 倍であった。クエチアピンは、体表面積 mg/m2 に基づいて MRHD 800 mg/日の約 1 倍の経口用量で、メスの Sprague-Dawley ラットの交尾と繁殖力に悪影響を及ぼしました。薬物関連の影響には、交尾回数の減少と妊娠に至る交配回数の減少、および交尾までの間隔の増加が含まれていました。不規則な発情周期の増加は、10 および 50 mg/kg の用量で観察され、mg/m2 体表面積に基づいて 800 mg/日の MRHD の約 0.1 倍および 1 倍でした。雌ラットにおける無影響用量は 1 mg/kg、または mg/m2 体表面積に基づいて 800 mg/日の MRHD の 0.01 倍でした。
特定の集団での使用
妊娠
妊娠暴露登録
妊娠中にセロクエル 50 mg を含む非定型抗精神病薬に曝露した女性の妊娠転帰を監視する妊娠曝露登録があります。医療提供者は、非定型抗精神病薬の国家妊娠登録に連絡して患者を登録することをお勧めします。 http://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/
リスクの概要
妊娠第三期に抗精神病薬(セロクエルを含む)にさらされた新生児は、分娩後に錐体外路症状および/または禁断症状のリスクがあります( 臨床上の考慮事項 )。クエチアピンに曝露された妊婦の公表された疫学研究から入手可能な全体的なデータは、主要な先天性欠損症、流産、または母体または胎児への有害転帰の薬物関連リスクを確立していません。 データ )。未治療の統合失調症、双極 I 型、または大うつ病性障害、および妊娠中のセロクエル 200 mg を含む抗精神病薬への暴露に関連する母親へのリスクがあります ( 臨床上の考慮事項 )。
動物実験では、ラットとウサギの両方で、800 mg/日の最大推奨ヒト用量 (MRHD) の約 1 倍と 2 倍での骨格骨化の遅延、および手根/足根屈曲の発生率の増加 (マイナーなMRHD の約 2 倍のウサギ胎児の軟部組織異常)。さらに、胎児の体重は両方の種で減少しました。母体毒性(体重の減少および/または死亡として観察される)は、ラットでは MRHD の 2 倍、ウサギでは MRHD の約 1~2 倍で発生した。
示された集団の主要な先天性欠損症および流産の推定背景リスクは不明です。すべての妊娠には、先天性欠損症、喪失、またはその他の有害な転帰の背景リスクがあります。米国の一般集団では、臨床的に認識された妊娠における主要な先天性欠損症および流産の推定背景リスクは、それぞれ 2 ~ 4% および 15 ~ 20% です。
臨床上の考慮事項
病気に関連する母体および/または胎児のリスク
未治療の統合失調症、または再発、入院、および自殺のリスクの増加を含む、双極 I 型障害による母親へのリスクがあります。統合失調症および双極 I 型障害は、早産を含む周産期の有害転帰の増加と関連しています。これが病気の直接の結果なのか、他の併存因子によるものなのかは不明です。
前向き縦断研究では、大うつ病性障害の病歴があり、妊娠初期に胸腺機能が正常で抗うつ薬を服用していた 201 人の妊婦を追跡しました。妊娠中に抗うつ薬を中止した女性は、抗うつ薬を継続した女性よりも大うつ病の再発を経験する可能性が高かった.妊娠中および産後に抗うつ薬による治療を中止または変更する場合は、未治療のうつ病のリスクを考慮してください。
胎児/新生児の有害反応
妊娠後期にセロクエル 50mg を含む抗精神病薬に曝露した新生児で、興奮、筋緊張亢進、筋緊張低下、振戦、傾眠、呼吸困難、および摂食障害を含む錐体外路症状および/または禁断症状が報告されています。これらの症状の重症度はさまざまでした。新生児の錐体外路および/または禁断症状を監視し、症状を適切に管理します。一部の新生児は、特別な治療を受けなくても数時間または数日で回復しました。他の人は長期の入院を必要としました。
データ
ヒューマンデータ
妊娠中の非定型抗精神病薬の使用に関する観察研究、出生登録、および症例報告から発表されたデータは、抗精神病薬および主要な先天異常との明確な関連を報告していません。妊娠中に抗精神病薬にさらされた 9,258 人の女性の Medicaid データベースからの後ろ向きコホート研究では、主要な先天性欠損症の全体的なリスクの増加は示されませんでした。
動物データ
妊娠中のラットとウサギが器官形成期にクエチアピンに曝露された場合、胎児に催奇形性の影響はありませんでした。用量は、ラットでは 25、50、および 200 mg/kg、ウサギでは 25、50、および 100 mg/kg で、MRHD の MRHD の約 0.3、0.6、および 2 倍 (ラット) および 0.6、1、および 2 倍 (ウサギ) でした。 mg/m2 体表面積に基づく 800 mg/日の統合失調症。しかし、ラットとウサギの両方で 800 mg/日の MRHD の約 1 倍と 2 倍で骨格骨化の遅延を含む胚-胎児毒性の証拠があり、手根骨/足根骨の屈曲 (軽度の軟部組織の異常) の発生率が増加した。 MRHD の約 2 倍のウサギ胎児。さらに、胎児の体重は両方の種で減少しました。母体毒性 (体重の減少および/または死亡として観察される) は、ラットでは MRHD の 2 倍、ウサギでは MRHD の約 1 ~ 2 倍 (試験したすべての用量) で発生しました。
ラットの周産期/出生後の生殖研究では、妊娠中の母動物に MRHD 800 mg/日の 0.01、0.1、および 0.2 倍の用量でクエチアピンを体表面積 mg/m2 に基づいて投与した場合、薬物関連の影響は観察されませんでした。しかし、予備的な周産期/出生後の研究では、MRHD の 3 倍で胎児と子犬の死亡が増加し、平均同腹体重が減少しました。
授乳
リスクの概要
公表された文献からの限られたデータでは、乳児の相対用量が母体の体重調整用量の 1% 未満であるヒト母乳中のクエチアピンの存在が報告されています。母乳を通じてクエチアピンにさらされた乳児で報告された一貫した有害事象はありません。牛乳生産に対するクエチアピンの影響に関する情報はありません。セロクエル 100mg に対する母親の臨床的必要性、およびセロクエルまたは母親の基礎疾患による母乳育児への潜在的な悪影響とともに、母乳育児の発達上および健康上の利点を考慮する必要があります。
生殖能力のある雌と雄
不妊
女性
クエチアピンの薬理学的作用 (D2 拮抗作用) に基づいて、セロクエルによる治療は血清プロラクチンレベルの上昇をもたらす可能性があり、生殖能力のある雌の生殖能力の可逆的な低下につながる可能性があります [参照 警告と注意事項 ]。
小児用
一般に、臨床試験中に小児および青年で観察された有害反応は、いくつかの例外を除いて、成人集団の有害反応と同様でした.収縮期および拡張期血圧の上昇は、小児および青年で発生し、成人では発生しませんでした。起立性低血圧は、小児および青年 ( 警告と注意事項 と 有害反応 ]。
統合失調症
13 歳から 17 歳の青少年の統合失調症治療におけるセロクエル 50mg の有効性と安全性は、1 つの 6 週間二重盲検プラセボ対照試験で実証されました。 適応症 、 投薬と管理 、 有害反応 、 と 臨床研究 ]。
統合失調症の 13 歳未満の小児患者におけるセロクエル 300mg の安全性と有効性は確立されていません。
メンテナンス
双極性障害の維持療法におけるセロクエルの安全性と有効性は、18 歳未満の小児患者では確立されていません。統合失調症の維持療法におけるセロクエルの安全性と有効性は、小児患者を含むどの患者集団においても確立されていません。
バイポーラマニア
10~17 歳の双極 I 型障害の小児および青年の躁病の治療におけるセロクエルの有効性と安全性は、3 週間の二重盲検プラセボ対照多施設試験で実証されました。 適応症 、 投薬と管理 、 有害反応 、 と 臨床研究 ]。双極性躁病の 10 歳未満の小児患者におけるセロクエルの安全性と有効性は確立されていません。
双極性うつ病
18 歳未満の双極性うつ病の小児患者におけるセロクエル 300mg の安全性と有効性は確立されていません。セロクエル XR の臨床試験は、双極性うつ病の小児および青年 (10~17 歳) を対象に実施されましたが、有効性は確立されていません。
クエチアピンの薬物動態には、小児/青年 (10 ~ 17 歳) と成人の間でいくつかの違いが見られました。体重で調整すると、クエチアピンの AUC と Cmax は、成人と比較して、小児と青年でそれぞれ 41% と 39% 低かった。活性代謝物であるノルクエチアピンの薬物動態は、体重調整後、小児/青年と成人の間で類似していた[ 臨床薬理学 ]。
高齢者の使用
セロクエル 300mg の臨床試験に参加した約 3,700 人の患者のうち、7% (232 人) が 65 歳以上でした。一般に、高齢者と若年成人とでセロクエルの忍容性が異なるという兆候はありませんでした。それでもなお、薬物動態学的クリアランスを低下させたり、セロクエル 200mg に対する薬力学的反応を高めたり、耐性や起立性を低下させたりする可能性のある要因が存在する場合は、開始用量を下げ、滴定を遅くし、初回投与期間中の注意深いモニタリングを検討する必要があります。お年寄り。セロクエル 25mg の平均血漿クリアランスは、若年患者と比較して、高齢患者では 30% から 50% 減少した [参照 臨床薬理学 と 投薬と管理 ]。
腎障害
腎障害のある患者におけるセロクエルの臨床経験は限られている[参照 臨床薬理学 ]。
肝障害
クエチアピンは肝臓で広範囲に代謝されるため、肝障害のある患者ではより高い血漿レベルが予想されます。この集団では、1 日 25 mg という低用量の開始が推奨され、1 日 25 mg から 50 mg の増分で用量を増やすことができます [参照 投薬と管理 と 臨床薬理学 ]。
過剰摂取
人間の経験
臨床試験では、最大30グラムのクエチアピンの急性過剰摂取で生存が報告されています.過剰摂取したほとんどの患者は、副作用を経験しなかったか、報告された事象から完全に回復しました. 13.6グラムのクエチアピン単独の過剰摂取後の臨床試験で死亡が報告されています.一般に、報告されている徴候や症状は、眠気、鎮静、頻脈、低血圧、昏睡やせん妄を含む抗コリン作動性毒性など、薬物の既知の薬理学的効果の誇張に起因するものでした。既存の重度の心血管疾患を持つ患者は、過剰摂取の影響のリスクが高くなる可能性があります[参照 警告と注意事項 ]。 9600 mg の推定過剰摂取を含む 1 つのケースは、低カリウム血症と第 1 度心臓ブロックに関連付けられました。市販後の経験では、過剰摂取によるQT延長の報告例がありました。
過剰摂取の管理
気道を確立して維持し、十分な酸素化と換気を確保します。心血管モニタリングを直ちに開始し、不整脈の可能性を検出するために継続的な心電図モニタリングを含める必要があります。
適切な支援策が管理の要です。セロクエル XR の過剰摂取の管理に関する最新情報については、認定された地域毒物管理センター (1-800-222-1222) にお問い合わせください。
禁忌
クエチアピンまたはセロクエル XR 製剤中の賦形剤に対する過敏症。セロクエル XR で治療された患者でアナフィラキシー反応が報告されています。
臨床薬理学
作用機序
列挙された適応症におけるクエチアピンの作用機序は不明である。ただし、これらの適応症におけるクエチアピンの有効性は、ドーパミン 2 型 (D2) とセロトニン 2 型 (5HT2) の拮抗作用の組み合わせによって媒介される可能性があります。活性代謝物である N-デスアルキル クエチアピン (ノルクエチアピン) は、親薬物 (クエチアピン) よりも D2 で同様の活性を示しますが、5HT2A 受容体でより大きな活性を示します。
薬力学
クエチアピンとその代謝産物であるノルクエチアピンは、複数の神経伝達物質受容体に親和性があり、ノルクエチアピンは一般にクエチアピンよりも高い親和性で結合します。ドーパミン D1 でのクエチアピンとノルクエチアピンの Ki 値は 428/99.8 nM、D2 で 626/489nM、セロトニン 5HT1A で 1040/191 nM 5HT2A で 38/2.9 nM、ヒスタミン H1 で 4.4/1.1 nM、ムスカリン M1 で 1086/それぞれ 38.3 nM、アドレナリン α1b では 14.6/46.4 nM、α2 受容体では 617/1290 nM。クエチアピンとノルクエチアピンには、ベンゾジアゼピン受容体に対するかなりの親和性がありません。
QT間隔への影響
臨床試験では、クエチアピンは QT 間隔の持続的な増加とは関連していませんでした。ただし、QT 効果は徹底的な QT 研究で体系的に評価されていません。市販後の経験では、クエチアピンを過剰摂取した患者で QT 延長が報告された例があった [ 過剰摂取 ]、併存疾患のある患者、および電解質の不均衡を引き起こしたり、QT間隔を増加させることが知られている薬を服用している患者.
薬物動態
大人
クエチアピン フマル酸活性は、主に親薬によるものです。クエチアピンの複数回投与の薬物動態は、提案された臨床用量範囲内で用量に比例し、クエチアピンの蓄積は複数回投与で予測可能です。クエチアピンの排除は、主に肝代謝によるものであり、提案された臨床用量範囲内で平均終末半減期は約 6 時間です。定常状態の濃度は、投与後 2 日以内に達成されると予想されます。クエチアピンは、シトクロム P450 酵素によって代謝される薬物の代謝を妨害する可能性は低いです。
子供と青年
定常状態では、親化合物の薬物動態は、小児および青年 (10 ~ 17 歳) において、成人と同様でした。しかし、用量と体重で調整すると、親化合物の AUC と Cmax は、成人よりも小児と青年でそれぞれ 41% と 39% 低かった。活性代謝物であるノルクエチアピン、AUC、および Cmax については、成人よりも小児および青年で、それぞれ 45% および 31% 高かった。用量と体重を調整すると、代謝産物であるノルクエチアピンの薬物動態は、小児と青年および成人の間で類似していた[参照 特定の集団での使用 ]。
吸収
クエチアピン フマル酸塩は、経口投与後急速に吸収され、1.5 時間で最高血漿濃度に達します。錠剤製剤は、溶液に対して 100% 生物学的に利用可能です。クエチアピンのバイオアベイラビリティは、食物と一緒に投与してもわずかに影響を受け、Cmax 値と AUC 値はそれぞれ 25% と 15% 増加します。
分布
クエチアピンは全身に広く分布しており、見かけの分布体積は 10±4 L/kg です。治療濃度で血漿タンパク質に 83% 結合します。 In vitro では、クエチアピンはワルファリンまたはジアゼパムのヒト血清アルブミンへの結合に影響を与えませんでした。同様に、ワルファリンもジアゼパムもクエチアピンの結合を変化させませんでした。
代謝と排泄
14C-クエチアピンの単回経口投与後、未変化体として排泄される量は投与量の 1% 未満であり、クエチアピンが高度に代謝されることを示しています。投与量の約 73% と 20% がそれぞれ尿と糞で回収されました。
クエチアピンは、肝臓で広範囲に代謝されます。主な代謝経路は、スルホキシド代謝物へのスルホキシド化と親酸代謝物への酸化です。両方の代謝産物は薬理学的に不活性です。ヒト肝ミクロソームを使用した in vitro 研究では、シトクロム P450 3A4 アイソザイムが、クエチアピンの主要であるが不活性なスルホキシド代謝物への代謝、およびその活性代謝物 N-脱アルキル クエチアピンの代謝に関与していることが明らかになりました。
年
クエチアピンの経口クリアランスは、若年患者 (n=12) と比較して高齢患者 (65 歳以上、n=9) では 40% 減少し、投与量の調整が必要になる場合がある [参照 投薬と管理 ]。
性別
クエチアピンの薬物動態に対する性別の影響はありません。
人種
クエチアピンの薬物動態に人種の影響はありません。
喫煙
喫煙は、クエチアピンの経口クリアランスに影響を与えません。
腎不全
重度の腎障害のある患者 (Clcr=10-30 mL/min/1.73 m2、n=8) は、正常な被験者 (Clcr > 80 mL/min/1.73 m2、n=8) よりも平均経口クリアランスが 25% 低かったが、腎不全の被験者の血漿クエチアピン濃度は、同じ用量を投与された正常な被験者で見られる濃度の範囲内でした。したがって、これらの患者では用量調整は必要ありません[ 特定の集団での使用 ]。
肝不全
肝障害のある患者 (n=8) は、正常な被験者よりもクエチアピンの平均経口クリアランスが 30% 低かった。 8 人の肝障害患者のうち 2 人では、AUC と Cmax が健康な被験者で通常観察される値の 3 倍でした。クエチアピンは肝臓で広範囲に代謝されるため、肝障害のある集団ではより高い血漿レベルが予想され、投与量の調整が必要になる場合があります。 投薬と管理 と 特定の集団での使用 ]。
薬物間相互作用研究
クエチアピンの薬物動態に対する他の薬物の効果の in vivo 評価を表 17 に要約する [ 投薬と管理 と 薬物相互作用 ]。
in vitro 酵素阻害データは、クエチアピンとその代謝産物のうちの 9 つは、シトクロム CYP 1A2、2C9、2C19、2D6、および 3A4 によって媒介される in vivo 代謝に対してほとんど阻害効果がないことを示唆しています。 750 mg/日の用量のクエチアピンは、アンチピリン、リチウム、またはロラゼパムの単回投与の薬物動態に影響を与えませんでした (表 18) [参照 薬物相互作用 ]。
動物毒物学および/または薬理学
クエチアピンは、4 週間以上のラット毒性試験およびマウスの 2 年間の発がん性試験で、甲状腺の色素沈着を用量依存的に増加させた。ラットの研究では、用量は 10、25、50、75、150、および 250 mg/kg であり、体表面積 mg/m2 に基づく MRHD 800 mg/日の約 0.1、0.3、0.6、1、2、および 3 倍です。 、 それぞれ。マウスの発がん性試験における用量は 20、75、250、および 750 mg/kg であり、体表面積 mg/m2 に基づく MRHD 800 mg/日の約 0.1、0.5、1.5、および 4.5 倍です。色素沈着は、ラットでは不可逆的であることが示されました。色素の正体は特定できませんでしたが、甲状腺濾胞上皮細胞でクエチアピンと共局在することがわかりました。機能的影響と、この所見のヒトへのリスクとの関連性は不明です。
クエチアピンを 6 か月または 12 か月投与された犬では、1 か月間ではなく、100 mg/kg の用量または 800 の MRHD の 4 倍の用量で、水晶体の外側皮質の後方縫合の接合部に限局性三角白内障が発生しました。 mg/m2 体表面積に基づく mg/日。この発見は、クエチアピンによるコレステロール生合成の阻害による可能性があります。クエチアピンは、反復投与のイヌおよびサルの研究で、血漿コレステロール値の用量依存的な低下を引き起こしました。しかし、血漿コレステロールと個々の犬の白内障の存在との間に相関関係はありませんでした。血漿中のデルタ-8コレスタノールの出現は、これらの種におけるコレステロール生合成の後期段階の阻害と一致しています。また、クエチアピンで治療された雌犬の特別な研究で観察されたレンズの外皮質のコレステロール含有量が25%減少しました.薬物関連の白内障は、他の種では見られません。しかし、サルでの 1 年間の研究では、225 mg/kg の用量または 800 mg/日の MRHD の 5.5 倍 (mg/m2 体に基づく) の用量で、2/7 のメスで水晶体前面の横紋状の外観が検出されました。表面積。
臨床研究
統合失調症
短期試験 - 成人
統合失調症の治療におけるセロクエル 50mg の有効性は、統合失調症の DSM III-R 基準を満たす統合失調症の入院患者を対象とした 3 つの短期 (6 週間) 対照試験で確立されました。 3 つの試験のうちの 1 つでは、ハロペリドール固定用量の単回投与群が比較治療として含まれていましたが、このハロペリドール単回投与群は、セロクエル 200mg とハロペリドールの信頼できる有効な比較を提供するには不十分でした。
これらの研究では、精神医学的徴候と症状を評価するためにいくつかの手段が使用されました。その中には、統合失調症の薬物治療の効果を評価するために伝統的に使用されてきた一般的な精神病理学の複数項目の目録である簡易精神医学評価尺度 (BPRS) があります。 BPRS 精神病クラスター (概念の混乱、幻覚行動、疑わしさ、異常な思考内容) は、活発な精神病の統合失調症患者を評価するための特に有用なサブセットと見なされます。 2 番目の従来の評価である臨床全体印象 (CGI) は、患者の全体的な臨床状態について、統合失調症の症状に十分に精通している熟練した観察者の印象を反映しています。
試行の結果は次のとおりです。
成人の統合失調症の治療におけるこれら 3 つの研究の主要な有効性の結果を表 19 に示します。
集団サブセット(人種、性別、および年齢)の検査では、人種または性別に基づく反応性の違いは明らかにされず、40 歳以上の患者と比較して 40 歳未満の患者で明らかに大きな影響が見られました。この所見は不明です。
青少年(13~17歳)
青少年(13~17 歳)の統合失調症の治療におけるセロクエル 300mg の有効性は、6 週間の二重盲検プラセボ対照試験(試験 4)で実証されました。統合失調症の DSM-IV 診断基準を満たした患者は、セロクエル 400 mg/日 (n=73)、セロクエル 800 mg/日 (n=74)、またはプラセボ (n=75) の 3 つの治療グループのいずれかに無作為に割り付けられました。治験薬は 50 mg/日から開始し、2 日目には 100 mg/日に増量しました (1 日 2 ~ 3 回に分けて投与)。続いて、1日2回または3回に分けて、1日100mg単位で1日400mgまたは800mgの目標用量まで用量を調整した。主要な有効性変数は、ポジティブおよびネガティブ シンドローム スケール (PANSS) のベースラインからの平均変化でした。
400 mg/日および 800 mg/日のセロクエルは、PANSS 合計スコアの低下においてプラセボよりも優れていました。青年期の統合失調症の治療におけるこの研究の主な有効性の結果を表 19 に示します。
双極性障害
双極Ⅰ型障害、躁病または混合エピソード
大人
躁病エピソードの急性治療におけるセロクエルの有効性は、躁病エピソードを伴うバイポーラ I 障害の DSM-IV 基準を満たす患者を対象とした 3 つのプラセボ対照試験で確立されました。これらの試験には、精神病的特徴の有無にかかわらず患者が含まれ、急速なサイクリングと混合エピソードのある患者は除外されました。これらの試験のうち、2 件は単剤療法 (12 週間) で、1 件はリチウムまたはジバルプロエックスの補助療法 (3 週間) でした。これらの試験の主要なアウトカムは、単剤療法の場合は 3 週間および 12 週間、補助療法の場合は 3 週間でのヤングマニア評価尺度(YMRS)スコアのベースラインからの変化でした。補助療法は、セロクエル 100mg とリチウムまたはジバルプロエックスの同時開始またはその後の投与と定義されます。
これらの試験で躁症状を評価するために使用された主要な評価手段はYMRSであり、躁症状の程度(過敏性、破壊的/攻撃的な行動、睡眠、気分の高揚、発話、活動の増加、性的関心、言語/思考障害、思考内容、外見、および洞察) を 0 (躁病の特徴なし) から 60 (最大スコア) の範囲で評価します。
試行の結果は次のとおりです。
単剤療法
双極性躁病の急性期治療におけるセロクエルの有効性は、2 つのプラセボ対照試験で確立されました。セロクエルとプラセボを比較した 2 つの 12 週間試験 (n=300, n=299) では、3 週目と 12 週目の YMRS 合計スコアの減少において、セロクエル 300mg がプラセボよりも優れていました。 400 mg/日から 800 mg/日の範囲で投与された (表 20 の研究 1 および 2)。
補助療法
この 3 週間のプラセボ対照試験では、双極性躁病 (YMRS ≥20) の 170 人の患者が無作為にセロクエル 100mg またはプラセボをリチウムまたはジバルプロエックスの補助療法として投与されました。患者は、無作為化の前にリチウムまたはジバルプロエックスの適切な治療コースを受けている場合と受けていない場合があります。セロクエル 50mg は、リチウムまたはジバルプロエックス単独に追加した場合、YMRS 合計スコアの低下においてプラセボよりも優れていました (表 20 の研究 3)。
セロクエル 25 mg を服用しているこの試験の患者の大部分は、1 日あたり 400 mg から 800 mg の範囲で投与されました。同様に設計された試験 (n=200) では、セロクエルは YMRS スコアの改善と関連していましたが、おそらくプラセボ効果が高かったため、プラセボに対する優位性は示されませんでした。
成人の躁病の治療におけるこれらの研究の主な有効性結果を表 20 に示します。
小児および青年(10~17 歳)
小児および青年(10~17 歳)の双極 I 型障害に関連する躁病エピソードの急性治療におけるセロクエルの有効性は、3 週間の二重盲検プラセボ対照多施設試験で実証されました(研究 4表 20)。躁病エピソードの DSM-IV 診断基準を満たした患者は、セロクエル 400 mg/日 (n=95)、セロクエル 600 mg/日 (n=98)、プラセボ (n=91) の 3 つの治療グループのいずれかに無作為に割り付けられました。 .治験薬は 50 mg/日で開始し、2 日目には 100 mg/日に増量しました (1 日 2 回または 3 回の分割投与)。その後、用量は、1 日 2 回または 3 回に分けて 1 日 100 mg ずつ増量して、目標用量の 400 mg/日または 600 mg/日になるように滴定されました。主な有効性変数は、合計 YMRS スコアのベースラインからの平均変化でした。
セロクエル 400 mg/日および 600 mg/日は、YMRS 合計スコアの低下においてプラセボよりも優れていました (表 20)。
双極性障害、うつ病エピソード
大人
双極性障害に伴う抑うつエピソードの急性期治療に対するセロクエル 25mg の有効性は、同じようにデザインされた 2 つの 8 週間無作為化二重盲検プラセボ対照試験 (N=1045) で確立されました (表 21 の試験 5 および 6)。 .これらの研究には、双極Ⅰ型障害または双極Ⅱ型障害のいずれかの患者と、急速なサイクリングコースがある患者とない患者が含まれていました。セロクエル 200 mg に無作為に割り付けられた患者には、300 mg または 600 mg の固定用量が 1 日 1 回投与されました。
これらの研究で抑うつ症状を評価するために使用された主要な評価手段は、0 から 60 までのスコアを持つ 10 項目の臨床医評価尺度である Montgomery-Asberg Depression Rating Scale (MADRS) でした。両方の研究の主要評価項目は、両方の研究で、セロクエル 50mg は MADRS スコアの低下においてプラセボよりも優れていました。プラセボと比較した MADRS スコアの変化によって測定される症状の改善は、8 日目 (1 週目) 以降の両方の研究で見られました。これらの研究では、600 mg の用量で追加の利点は見られませんでした。 Q-LES-Q(SF)を使用して測定した場合、300 mg 投与群では、プラセボよりも統計的に有意な改善が、全体的な QOL および機能のさまざまな領域に関連する満足度で見られました。
成人の双極性障害に関連する抑うつエピソードの急性治療におけるこれらの研究の主要な有効性の結果を表 21 に示します。
リチウムまたはDivalproexの補助としての維持治療
双極 I 型障害の維持治療におけるセロクエル 100mg の有効性は、双極 I 型障害の DSM-IV 基準を満たす患者 (n=1326) を対象とした 2 つのプラセボ対照試験で確立されました (図 1 および 2 の試験 7 および 8)。試験には、最新のエピソードが躁病、抑うつ、または混合であり、精神病の特徴の有無にかかわらず患者が含まれていました。非盲検段階では、無作為化するために、患者は少なくとも 12 週間セロクエルとリチウムまたは divalproex で安定している必要がありました。平均して、患者は 15 週間安定していました。無作為化フェーズでは、患者はリチウムまたはジバルプロエックスによる治療を継続し、セロクエル (1 日 2 回、合計 400 mg/日から 800 mg/日を投与) またはプラセボのいずれかを投与するよう無作為に割り付けられました。患者の約 50% が 280 日目までにセロクエル群を中止し、プラセボ群の 50% が二重盲検治療の 117 日目までに中止しました。これらの研究の主要評価項目は、気分イベント (躁病、混合、または抑うつエピソード) の再発までの時間でした。気分イベントは、気分エピソードの投薬開始または入院として定義されました。 -2回連続の評価でYMRSスコア≧20またはMADRSスコア≧20;または気分イベントによる研究の中止 (図 1 および図 2)。
両方の研究で、セロクエルは気分イベントの再発までの時間を延長する点でプラセボよりも優れていました。治療効果は、躁病エピソードとうつ病エピソードの両方の再発までの時間の増加に見られました。セロクエルの効果は、特定のサブグループ(気分安定剤、性別、年齢、人種、最近の双極性エピソード、急速サイクリングコースに割り当てられたもの)とは無関係でした。
図 1: 気分イベントの再発までの時間のカプラン・マイヤー曲線 (研究 7)
図 2: 気分イベントの再発までの時間のカプラン・マイヤー曲線 (研究 8)
患者情報
セロクエル (SER-oh-kwell)(フマル酸クエチアピン)錠
セロクエルの服用を開始する前と、リフィルを入手するたびに、この投薬ガイドをお読みください。新しい情報があるかもしれません。この情報は、あなたの病状や治療について医療提供者と話すことに代わるものではありません.
セロクエルについて知っておくべき最も重要な情報は何ですか?
セロクエル 100mg は、次のような重大な副作用を引き起こす可能性があります。
- 以下について、あなたまたはあなたの家族の医療提供者に相談してください。
- 抗うつ薬による治療のすべてのリスクと利点。
- うつ病やその他の深刻な精神疾患に対するすべての治療法の選択肢
- 抗うつ薬は、治療の最初の数か月以内に、一部の子供、10代の若者、および若い成人で自殺念慮または行動を増加させる可能性があります.
- うつ病やその他の深刻な精神疾患は、自殺念慮や自殺行動の最も重要な原因です。一部の人々は、自殺念慮または行動を起こすリスクが特に高い可能性があります。 これらには、うつ病、双極性障害(躁うつ病とも呼ばれる)、または自殺念慮または行動を起こしている(またはその家族歴がある)人が含まれます。
- 自分や家族の自殺念慮や自殺行為を監視し、防止するにはどうすればよいですか?
- 気分、行動、思考、または感情の変化、特に突然の変化に細心の注意を払ってください。これは、抗うつ薬の服用を開始するとき、または用量を変更するときに非常に重要です。
- すぐに医療提供者に電話して、気分、行動、思考、または感情の新しい変化または突然の変化を報告してください。
- 医療提供者とのすべてのフォローアップ訪問は、予定どおりに行ってください。特に症状について懸念がある場合は、必要に応じて訪問の合間に医療提供者に電話してください。
あなたまたはあなたの家族に次のような症状がある場合、特に新しい症状、悪化した症状、心配な症状がある場合は、すぐに医療提供者に連絡してください。
- 自殺や死についての考え
- 自殺しようとする
- 新しいまたは悪化したうつ病
- 新しいまたは悪化した不安
- 非常に動揺している、または落ち着きがない
- パニック発作
- 寝つきが悪い(不眠症)
- 新しいまたは悪化した過敏症
- 攻撃的、怒っている、または暴力的である
- 危険な衝動で行動する
- 活動と会話の極端な増加 (躁病)
- 行動や気分のその他の異常な変化
抗うつ薬について他に何を知る必要がありますか?
- 最初に医療提供者に相談することなく、抗うつ薬を中止しないでください。 抗うつ薬を突然中止すると、他の症状を引き起こす可能性があります。
- 抗うつ薬は、うつ病やその他の病気の治療に使用される薬です。 うつ病を治療するリスクと、治療しないリスクについて話し合うことが重要です。患者とその家族、または他の介護者は、抗うつ薬の使用だけでなく、すべての治療法について医療提供者と話し合う必要があります。
- 抗うつ薬には他の副作用があります。 あなたまたはあなたの家族に処方された薬の副作用について、医療提供者に相談してください。
- 抗うつ薬は他の薬と相互作用することがあります。 あなたやあなたの家族が服用しているすべての薬を知っておいてください。医療提供者に見せるために、すべての薬のリストを保管してください。最初に医療提供者に確認することなく、新しい薬を開始しないでください。
- 子供に処方されるすべての抗うつ薬が、子供への使用が FDA に承認されているわけではありません。 詳細については、お子様の医療提供者にご相談ください。
セロクエル300mgとは?
セロクエル 100mg は、以下の治療に使用される処方薬です。
- 13歳以上の統合失調症
- 以下を含む成人の双極性障害:
- 双極性障害に関連する抑うつエピソード
- 双極I型障害単独またはリチウムまたはdivalproexに関連する躁病エピソード
- リチウムまたはジバルプロエックスによる双極I型障害の長期治療
- 10~17歳の小児における双極I型障害に関連する躁病エピソード
セロクエル 100mg が 10 歳未満の子供に安全で有効であるかどうかはわかっていません。
セロクエル 100mg を服用する前に医療提供者に何を伝えるべきですか?
セロクエル 200mg を服用する前に、次のことを持っているか、または持っていたかどうかを医療提供者に伝えてください。
- あなたまたはあなたの家族の糖尿病または高血糖。かかりつけの医療提供者は、セロクエル 300mg を開始する前、および治療中に血糖値をチェックする必要があります。
- 高レベルの総コレステロール、トリグリセリドまたは LDL コレステロール、または低レベルの HDL コレステロール
- 低血圧または高血圧
- 低白血球数
- 白内障
- 発作
- 異常な甲状腺検査
- 高プロラクチン値
- 心の問題
- 肝臓の問題
- その他の病状
- 妊娠中または妊娠予定の方。セロクエル 100mg が胎児に害を及ぼすかどうかはわかっていません。
- セロクエルの投与中に妊娠した場合は、かかりつけの医療提供者に、非定型抗精神病薬の国家妊娠登録簿への登録について相談してください。 1-866-961-2388 に電話して登録するか、 http://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/
- 授乳中または授乳予定。セロクエルは母乳に移行する可能性があります。セロクエルを投与された場合の最善の授乳方法については、担当の医療提供者に相談してください。
- 膀胱を完全に空にすることができない状態 (尿閉)、前立腺肥大、便秘、または眼圧の上昇がある場合。
あなたが服用している、または最近服用したすべての薬について医療提供者に伝えてください 処方薬、市販薬、ハーブサプリメント、ビタミンなど。
セロクエル 300mg と他の薬は互いに影響し合い、重大な副作用を引き起こす可能性があります。セロクエル 200mg は他の薬の作用に影響を与える可能性があり、他の薬はセロクエル 200mg の作用に影響を与える可能性があります。
セロクエルが検査結果に影響を与える可能性があるため、尿中薬物検査を受けている場合は、医療提供者に伝えてください。セロクエルを服用していることを検査者に伝えてください。
セロクエルはどのように服用すればよいですか?
- セロクエルは、担当の医療提供者の指示どおりに服用してください。自分で用量を変更しないでください。
- セロクエル 25mg は、食事の有無にかかわらず経口で服用してください。
- セロクエル 25mg を中止する必要があると思われる場合は、まず医療提供者に相談してください。 セロクエルの服用を急に中止すると、寝つきが悪い、寝つきが悪い(不眠)、吐き気、嘔吐などの副作用があらわれることがあります。
- セロクエル 25mg を飲み忘れた場合は、気がついたらすぐに服用してください。次の服用が近づいている場合は、飲み忘れた分は飛ばしてください。通常の時間に次の用量を服用してください。医療提供者の指示がない限り、同時に 2 回分を服用しないでください。投与量がわからない場合は、医療提供者に電話してください。
セロクエル 100mg を服用している間、何を避けるべきですか?
- セロクエルがどのように影響するかを理解するまでは、運転、機械の操作、またはその他の危険な活動をしないでください。セロクエルは眠くなることがあります。
- 過熱や脱水を避ける。
- 過度に運動しないでください。
- 暑いときは、なるべく涼しいところに置いてください。
- 太陽を避けてください。厚着や厚着をしないでください。
- たくさん水を飲む。
- セロクエルの服用中は飲酒しないでください。セロクエルの一部の副作用を悪化させる可能性があります。
セロクエル 300mg の副作用にはどのようなものがありますか?
セロクエル 200mg は、次のような重大な副作用を引き起こす可能性があります。
- 「セロクエル 200mg について知っておくべき最も重要な情報は何ですか?」を参照してください。
- セロクエルのような薬を服用している認知症の高齢者では、死に至る可能性のある脳卒中が発生する可能性があります
- 悪性症候群(NMS)。 NMS は、セロクエルを含む抗精神病薬を服用している人に発生する可能性がある、まれではあるが非常に深刻な状態です。 NMS は死に至る可能性があり、病院で治療する必要があります。重度の病気になり、これらの症状の一部またはすべてがある場合は、すぐに医療提供者に連絡してください。
- 高熱
- 過度の発汗
- 硬い筋肉
- 錯乱
- 呼吸、心拍、血圧の変化
- 落ちる セロクエルを服用している一部の人に起こる可能性があります。これらの落下により、重傷を負う可能性があります。
- 高血糖(高血糖)。 すでに糖尿病にかかっている場合でも、糖尿病にかかったことがない場合でも、高血糖になる可能性があります。高血糖につながる可能性があります:
- ケトンによる血液中の酸の蓄積(ケトアシドーシス)
- 昏睡
- 死
セロクエルを服用している一部の人では、血糖値の上昇が起こることがあります。極端に高い血糖値は、昏睡または死に至る可能性があります。糖尿病または糖尿病の危険因子 (太りすぎや糖尿病の家族歴など) がある場合は、セロクエル 50mg を開始する前と治療中に、医療提供者が血糖値をチェックする必要があります。セロクエルの服用中に以下の高血糖(高血糖)の症状がみられた場合は、医療提供者に連絡してください。
- 非常にのどが渇く
- いつもより多く排尿する必要がある
- とてもお腹がすいた
- 脱力感や疲れを感じる
- 胃が痛い
- 混乱したり、息がフルーティーなにおいがしたりします
- 血中の脂肪レベルが高い(コレステロールとトリグリセリドの増加)。 セロクエルで治療を受けた人では、脂肪レベルが高くなることがあります。症状がない場合もあるため、担当の医療提供者は、セロクエルによる治療中にコレステロールとトリグリセリドをチェックすることを決定する場合があります.
- 体重増加(体重増加)。 セロクエルを服用している人は体重増加がよく見られるため、あなたと担当の医療提供者は定期的に体重をチェックする必要があります。健康的でバランスの取れた食事や運動など、体重増加を抑える方法について、担当の医療提供者に相談してください。
- 顔、舌、または他の体の部分で制御できない動き (遅発性ジスキネジー)。 これらは深刻な状態の兆候である可能性があります。遅発性ジスキネジアは、セロクエルの服用を中止しても治らない場合があります。セロクエルの服用を中止した後も、遅発性ジスキネジアが始まることがあります。
- 血圧の低下(起立性低血圧)、 座位または横臥位から急に立ち上がったときの心拍数と血圧の急激な変化によって引き起こされる立ちくらみまたは失神を含みます。
- 小児および 10 代の若者の血圧の上昇。 かかりつけの医療提供者は、セロクエルを開始する前および治療中に小児および青年の血圧をチェックする必要があります。
- 白血球数が少ない。 発熱、インフルエンザのような症状、またはその他の感染症がある場合は、白血球数が非常に少ないことが原因である可能性があるため、できるだけ早く医療機関に連絡してください.かかりつけの医療提供者は、白血球レベルをチェックして、さらなる治療やその他の処置が必要かどうかを判断する場合があります。
- 白内障
- 発作
- 異常な甲状腺検査。 医療提供者は、甲状腺ホルモンのレベルを調べるために血液検査を行うことがあります。
- プロラクチンレベルの上昇
- 眠気、眠気、疲労感、思考困難、通常の活動が困難
- 体温上昇
- 嚥下困難
- セロクエルの服用を突然中止すると、睡眠障害または睡眠障害(不眠症)、吐き気、または嘔吐. これらの症状は通常、症状が出始めてから 1 週間後に改善します。
セロクエル 50mg の最も一般的な副作用は次のとおりです。
大人の場合:
- 眠気
- 立っていると血圧が急激に下がる
- 体重の増加
- 鈍さ
- 異常な肝臓検査
- 胃のむかつき
- 口渇
- めまい
- 弱点
- 腹痛
- 便秘
- 喉の痛み
小児および青年では:
- 眠気
- めまい
- 倦怠感
- 吐き気
- 口渇
- 体重の増加
- 食欲増進
- 嘔吐
- 速い心拍
これらは、セロクエルの考えられるすべての副作用ではありません。詳細については、医療提供者または薬剤師にお尋ねください。
副作用に関する医学的アドバイスについては、医師に連絡してください。 1-800-FDA-1088 で副作用を FDA に報告できます。
セロクエル 100mg の保存方法は?
- セロクエル 100mg は室温、68°F ~ 77°F (20°C ~ 25°C) で保管してください。
- セロクエルとすべての医薬品は子供の手の届かないところに保管してください。
セロクエルの安全で効果的な使用に関する一般情報
医薬品は、医薬品ガイドに記載されている以外の目的で処方されることがあります。セロクエルが処方されていない状態には使用しないでください。他の人があなたと同じ症状であっても、セロクエルを与えないでください。それらに害を及ぼす可能性があります。
この投薬ガイドは、セロクエルに関する最も重要な情報をまとめたものです。さらに詳しい情報が必要な場合は、医療提供者に相談してください。医療専門家向けに書かれたセロクエル 100mg に関する情報については、薬剤師または医療提供者にお尋ねください。
詳細については、 www.セロクエル.com 、または 1-800-236-9933 までお電話ください。
セロクエル200mgの成分は?
有効成分: クエチアピンフマル酸塩
不活性成分: ポビドン、二塩基性リン酸二カルシウム二水和物、微結晶性セルロース、デンプングリコール酸ナトリウム、ラクトース一水和物、ステアリン酸マグネシウム、ヒプロメロース、ポリエチレングリコール、および二酸化チタン。 25 mg の錠剤には、赤と黄色の酸化鉄が含まれています。 100 mg と 400 mg の錠剤には、黄色三二酸化鉄のみが含まれています。
この医薬品ガイドは、米国食品医薬品局によって承認されています。