Rebetol 200mg Ribavirin 使用法、副作用および投与量。 オンライン薬局の価格。 処方箋不要のジェネリック医薬品。
Rebetol 200mg とは何ですか?
Rebetol は慢性 C 型肝炎の症状を治療するために使用される処方薬です。
Rebetol は、B 型肝炎/C 型肝炎薬と呼ばれる薬のクラスに属しています。 RSV エージェント。
Rebetol 200mg が 3 歳未満の子供に安全で効果的かどうかはわかっていません。
Rebetol の副作用の可能性は何ですか?
Rebetol は、次のような重大な副作用を引き起こす可能性があります。
- 蕁麻疹、
- 呼吸困難、
- 顔、唇、舌、喉の腫れ、
- 視力の問題
- 上腹部の激しい痛みが背中に広がり、
- 吐き気、
- 嘔吐、
- 下痢、
- 新しいまたは悪化した咳、
- 熱、
- 刺すような胸の痛み、
- 喘鳴、
- 呼吸困難、
- 憂鬱症、
- 自傷行為の考え、
- 他人を傷つける考え、
- 肌が白くなったり、黄ばんだり、
- 濃い色の尿、
- 錯乱、
- 弱点、
- 寒気、
- 風邪のような症状、
- 腫れた歯茎、
- 口内炎、
- 皮膚のただれ、
- あざができやすい、
- 不正出血と、
- 立ちくらみ
- 上記の症状がある場合は、すぐに医療機関を受診してください。
- Rebetol の最も一般的な副作用は次のとおりです。
- 吐き気、
- 嘔吐、
- 食欲減少、
- 熱、
- 寒気、
- 揺れ、
- 低血球数、
- 貧血、
- 弱点、
- 疲れ、
- 頭痛、
- 筋肉痛、
- 気分の変化、
- 不安、そして
- 過敏性
気になる副作用や治らない副作用がある場合は、医師に相談してください。
これらは、Rebetol の考えられるすべての副作用ではありません。詳細については、医師または薬剤師にお尋ねください。
副作用に関する医学的アドバイスについては、医師に連絡してください。 1-800-FDA-1088 で副作用を FDA に報告できます。
警告
重篤な疾患およびリバビリン関連作用のリスク
- REBETOL 単独療法は慢性 C 型肝炎ウイルス感染症の治療には効果がなく、この適応症に対して単独で使用すべきではありません [警告と注意事項を参照]。
- リバビリンの主な毒性は溶血性貧血です。 REBETOL 200mg 療法に関連する貧血は、致命的および非致命的な心筋梗塞を引き起こした心疾患の悪化をもたらす可能性があります。重度または不安定な心疾患の既往歴のある患者は、REBETOL で治療すべきではありません [用法・用量、警告と予防措置、および副作用を参照]。
- リバビリンに暴露されたすべての動物種で、重大な催奇形性および殺胚性効果が実証されています。さらに、リバビリンの複数回投与による半減期は 12 日間であるため、非血漿コンパートメントでは 6 か月も持続する可能性があります。したがって、REBETOL 療法は、妊娠中の女性および妊娠中の女性の男性パートナーには禁忌です。 REBETOL 療法を受けている女性患者および男性患者の女性パートナーの両方において、治療中および治療終了後 6 か月間は、妊娠を避けるために細心の注意を払う必要があります。治療中および治療後 6 か月のフォローアップ期間中に、少なくとも 2 つの信頼できる有効な避妊法を使用する必要があります [禁忌、警告および注意事項、特定集団での使用、非臨床毒性学、および患者情報を参照]。
説明
REBETOL (リバビリン) は、合成ヌクレオシド類似体 (プリン類似体) です。リバビリンの化学名は 1-β-D-リボフラノシル-1H-1,2,4-トリアゾール-3-カルボキサミドで、次の構造式を持っています (図 1 参照)。
図 1: 構造式
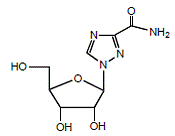
リバビリンは白色の結晶性粉末です。水に溶けやすく、無水アルコールに溶けにくい。実験式は C8H12N4O5 で、分子量は 244.21 です。
REBETOL カプセルは、白色の不透明なゼラチン カプセルに白色の粉末が入っています。各カプセルには 200 mg のリバビリンと、不活性成分である微結晶性セルロース、ラクトース一水和物、クロスカルメロース ナトリウム、およびステアリン酸マグネシウムが含まれています。カプセルシェルは、ゼラチン、ラウリル硫酸ナトリウム、二酸化ケイ素、および二酸化チタンで構成されています。カプセルは、シェラック、無水エチル アルコール、イソプロピル アルコール、n-ブチル アルコール、プロピレン グリコール、水酸化アンモニウム、および FD&C ブルー #2 アルミニウム レーキで作られた食用の青い医薬品インクで印刷されています。
レベトール 200mg 内服液は、無色透明から淡黄色のバブルガム風味の液体です。溶液 1 ミリリットルあたり 40 mg のリバビリンと、不活性成分のスクロース、グリセリン、ソルビトール、プロピレングリコール、クエン酸ナトリウム、クエン酸、安息香酸ナトリウム、風船ガム #15864 の天然および人工香料、および水が含まれています。
適応症
C型慢性肝炎(CHC)
REBETOL® (リバビリン) とインターフェロン α-2b (ペグ化および非ペグ化) の併用は、代償性肝疾患を有する 3 歳以上の患者の慢性 C 型肝炎 (CHC) の治療に適応されます [参照 警告と注意事項 、 と 特定の集団での使用 ]。
PegIntron® または INTRON A® との REBETOL 併用療法を開始する際には、次の点を考慮する必要があります。
- これらの適応症は、24 週間または 48 週間の治療後に検出不能な HCV-RNA を達成し、最後の投与から 24 週間後に持続的ウイルス学的反応 (SVR) を維持することに基づいています。
- REBETOL/PegIntron との併用療法は、REBETOL/INTRON A よりも好まれます。 臨床研究 ]。
- 以下の特徴を持つ患者は、一連の治療に失敗した後の再治療の恩恵を受ける可能性が低くなります:以前の無反応、以前のペグ化インターフェロン治療、重大な架橋線維症または肝硬変、および遺伝子型1感染[参照] 臨床研究 ]。
- 1年以上の治療に関する安全性と有効性のデータはありません。
投薬と管理
いかなる状況においても、レベトール カプセルを開けたり、つぶしたり、壊したりしないでください。 REBETOL 200mg は食事と一緒に摂取する必要があります。 臨床薬理学 ]。 REBETOL は、クレアチニンクリアランスが 50 mL/min 未満の患者には使用しないでください。
REBETOL/PegIntron 併用療法
成人患者
PegIntron の推奨用量は、患者の体重に基づいて、経口で 800 ~ 1400 mg の REBETOL 200 mg カプセルと組み合わせて、皮下に 1.5 mcg/kg/週です (表 1 を参照)。注射する PegIntron の量は、PegIntron の強度と患者の体重によって異なります。追加の投与情報については、PegIntron のラベルを参照してください。
治療期間 – インターフェロン α 未投与患者
遺伝子型 1 の患者の治療期間は 48 週間です。 12 週間の時点で HCV-RNA が少なくとも 2 log10 低下または消失していない患者、または治療の 24 週間後も HCV-RNA が検出可能なままである場合は、治療の中止を検討する必要があります。遺伝子型 2 および 3 の患者は、24 週間治療する必要があります。
治療期間 – 以前の治療失敗の PegIntron/REBETOL による再治療
以前に治療に失敗した患者の治療期間は、HCV 遺伝子型に関係なく 48 週間です。治療の 12 週目に HCV-RNA が検出されない患者、または治療の 24 週後も HCV-RNA が検出可能なままである再治療を受けた患者は、SVR を達成する可能性が非常に低く、治療の中止を検討する必要があります。 臨床研究 ]。
小児患者
小児患者の投与量は、PegIntron では体表面積によって、REBETOL では体重によって決定されます。 PegIntron の推奨用量は、3 ~ 17 歳の小児患者に 60 mcg/m²/週を皮下投与し、1 日 15 mg/kg/日の REBETOL 200 mg を 2 回に分けて経口投与することです (表 2 を参照)。 PegIntron/REBETOL 200mg を投与されている間に 18 歳の誕生日を迎えた患者は、小児用投与レジメンを継続する必要があります。遺伝子型 1 の患者の治療期間は 48 週間です。遺伝子型 2 および 3 の患者は、24 週間治療する必要があります。
レベトール/イントロンA併用療法
大人
治療期間 – インターフェロン α 未投与患者
イントロン A の推奨用量は、300 万 IU を週 3 回皮下投与することです。レベトール カプセルの推奨用量は、患者の体重によって異なります (表 3 を参照)。以前にインターフェロンで治療されていない患者の推奨される治療期間は、24 ~ 48 週間です。治療期間は、ベースラインの疾患特性、治療への反応、およびレジメンの忍容性に応じて、患者ごとに個別化する必要があります [参照 適応症と使用法 、 有害反応 、 と 臨床研究 ]。 24 週間の治療後、ウイルス学的反応を評価する必要があります。 HCV-RNA が 24 週間までにアッセイの検出限界を下回らなかった患者では、治療の中止を考慮する必要があります。以前に治療を受けていない患者集団における 48 週間を超える治療に関する安全性と有効性のデータはありません。
治療期間 – 再発患者における INTRON A/REBETOL による再治療
非ペグ化インターフェロン単剤療法後に再発した患者では、推奨される治療期間は 24 週間です。
小児科
REBETOL の推奨用量は、経口で 1 日 15 mg/kg です (午前と午後の分割用量)。 INTRON A と組み合わせた REBETOL の小児投与量については、表 2 を参照してください。注射用の INTRON A は、体重 25 kg ~ 61 kg で、週 3 回皮下に 300 万 IU/m² です。体重が 61 kg を超える場合は、成人の用量表を参照してください。
遺伝子型 1 の小児患者の推奨治療期間は 48 週間です。24 週間の治療後、ウイルス学的反応を評価する必要があります。この時点までにHCV-RNAがアッセイの検出限界を下回っていない患者では、治療の中止を考慮する必要があります。遺伝子型 2/3 の小児患者の推奨治療期間は 24 週間です。
臨床検査
REBETOL 200mg で治療を受けたすべての患者に対して、治療開始前とその後定期的に以下の臨床検査を行うことをお勧めします。
標準的な血液学的検査 - ヘモグロビンを含む (治療前、治療の 2 週目と 4 週目、および臨床的に適切な [参照] 警告と注意事項 ]、完全および分画白血球数、および血小板数。
- 血液化学 - 肝機能検査と TSH。
- 妊娠 - 出産の可能性がある女性の毎月のモニタリングを含む。
- 心電図 [参照 警告と注意事項 ]。
用量変更
REBETOL/INTRON A 併用療法または REBETOL/PegIntron 療法中に重度の副作用または臨床検査値の異常が発生した場合、副作用が弱まるか重症度が低下するまで用量を変更または中止する [参照 警告と注意事項 ]。用量調整後も不耐性が続く場合は、併用療法を中止する必要があります。 REBETOL/PegIntron 併用療法を受けている成人患者における PegIntron の減量は、最初の開始用量である 1.5 mcg/kg/週から 1 mcg/kg/週、そして 0.5 mcg/kg/週への 2 段階のプロセスで達成されます。 、 必要に応じて。 PegIntron の減量に関する追加情報については、PegIntron のラベルを参照してください。
成人併用療法の研究 2 では、体重 60 kg 以下の被験者の 57% を含む、PegIntron 1.5 mcg/kg と REBETOL 800 mg を毎日投与された被験者の 42% で用量が減少しました。研究 4 では、被験者の 16% が REBETOL と組み合わせて PegIntron を 1 mcg/kg に減量し、さらに 4% は有害事象のために PegIntron を 0.5 mcg/kg に 2 回減量する必要がありました [参照 有害反応 ]。
小児患者の減量は、PegIntron の推奨用量を 60 mcg/m²/週の初期開始用量から 40 mcg/m²/週、次に 20 mcg/m²/週に 2 段階のプロセスで変更することによって達成されます。必要です (表 4 を参照)。小児併用療法の試験では、PegIntron 60 mcg/m² を毎週、REBETOL 15 mg/kg を毎日投与された被験者の 25% で用量が減少しました。小児患者の減量は、レベトールの推奨用量を元の開始用量である 1 日 15 mg/kg から 2 段階で 12 mg/kg/日に変更し、必要に応じて 8 mg/kg/日に変更することによって達成されます (表 4 を参照してください)。
レベトール 200mg は、クレアチニンクリアランスが 50 mL/min 未満の患者には使用しないでください。腎機能障害のある患者および 50 歳以上の患者は、貧血の発生に関して注意深く監視する必要があります。 警告と注意事項 、 特定の集団での使用 、 と 臨床薬理学 ]。
REBETOL 200mg は、心疾患の既往がある患者には慎重に投与する必要があります。治療開始前に患者を評価し、治療中は適切にモニタリングする必要があります。心血管状態の悪化がある場合は、治療を中止する必要があります[参照 警告と注意事項 ]。
安定した心血管疾患の既往歴のある患者では、ヘモグロビンが 4 週間で 2 g/dL 以上減少した場合、永久的な減量が必要です。さらに、これらの心臓病歴のある患者の場合、減量して 4 週間後にヘモグロビンが 12 g/dL 未満のままである場合、患者は併用療法を中止する必要があります。
表 4 [ 警告と注意事項 ]。
INTRON A または PegIntron の用量を減らす方法の詳細については、INTRON A または PegIntron のラベルを参照してください。
投薬の中止
大人
PegIntron とリバビリンの併用療法を受けているインターフェロン アルファ未治療の HCV 遺伝子型 1 患者では、治療開始 12 週間で HCV-RNA の少なくとも 2 log10 の低下または喪失がない場合、または HCV-RNA がレベルは、治療の 24 週間後も検出可能なままです。遺伝子型に関係なく、12 週または 24 週の時点で HCV-RNA が検出された治療歴のある患者は、SVR を達成する可能性が非常に低く、治療の中止を検討する必要があります。
小児科(3~17歳)
PegIntron/REBETOL の組み合わせ(HCV ジェノタイプ 2 および 3 を除く)を受けている患者は、12 週目の HCV-RNA の低下が治療前と比較して 2 log10 未満の場合は 12 週目で、または 24 週目で検出可能な場合は治療を中止することをお勧めします。治療24週目のHCV-RNA。
供給方法
剤形と強度
レベトール 200mg カプセル 200mg
レベトール 200mg 内用液 40mg/mL
保管と取り扱い
レベトール200mgカプセル REBETOL 200 mg を含む白色の不透明なカプセルで、カプセル シェルに Schering Corporation のロゴが刻印されています。カプセルは56個のカプセルを含むボトルに包装されています( NDC 0085-1351-05)、70カプセル( NDC 0085-1385-07)、84カプセル( NDC 0085-1194-03)。
レベトール200mg内用液40mg 1 mL あたり、透明、無色から淡いまたは淡い黄色の風船ガム風味の液体で、4 オンスの琥珀色のガラス ボトル (100 mL/ボトル) に詰められ、チャイルド レジスタント クロージャー ( NDC 0085-1318-01)。
レベトール カプセルのボトルは 25°C (77°F) で保管してください。 15 ~ 30°C (59 ~ 86°F) まで許容されるエクスカーション [参照 USP制御の室温 ]。
REBETOL 200mg 経口溶液は、2 ~ 8°C (36 ~ 46°F) または 25°C (77°F) で保存する必要があります。 15 ~ 30°C (59 ~ 86°F) まで許容されるエクスカーション [参照 USP制御の室温 ]。
REBETOL 200mg Oral Solution は、米国ニュージャージー州 08889 の Whitehouse Station の子会社である Merck Sharp & Dohme Corp. のために製造されました。製造元: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Canada REBETOL カプセル製造元: Merck Sharp & Dohme Corp., Whitehouse Station, NJ 08889, USA の子会社。改訂: 2014 年 12 月。
副作用
PegIntron または INTRON A と組み合わせた REBETOL の臨床試験は、3 歳から 76 歳までの 7800 人を超える被験者で実施されています。
リバビリンの主な毒性は溶血性貧血です。ヘモグロビンレベルの低下は、経口療法の最初の 1 ~ 2 週間以内に発生しました。貧血に関連する心臓および肺の反応は、患者の約 10% で発生しました [ 警告と注意事項 ]。
臨床試験の全被験者の 96% 以上が 1 つ以上の副作用を経験しました。 PegIntron または INTRON A を REBETOL 200mg と組み合わせて投与された成人被験者で最も一般的に報告された副作用は、注射部位の炎症/反応、疲労/無力症、頭痛、硬直、発熱、吐き気、筋肉痛、および不安/情緒不安定/過敏性でした。 PegIntron または INTRON A と組み合わせて REBETOL 200mg を投与された 3 歳以上の小児被験者における最も一般的な副作用は、発熱、頭痛、好中球減少症、疲労、食欲不振、注射部位の紅斑、および嘔吐でした。
副作用のセクションでは、次の臨床試験を参照しています。
- REBETOL/PegIntron 併用療法試験:
- 臨床試験 1 – 評価された PegIntron 単剤療法 (このラベルにはこれ以上記載されていません。この試験に関する情報については、PegIntron のラベルを参照してください)。
- 研究 2 – 1.5 mcg/kg/週の PegIntron または INTRON A と組み合わせた REBETOL 800 mg/日の均一用量を評価しました。
- 試験 3 – PegIntron/体重ベースの REBETOL 200mg と PegIntron/一定用量の REBETOL 200mg レジメンを組み合わせて評価しました。
- 研究 4 – 2 つの PegIntron (1.5 mcg/kg/週および 1 mcg/kg/週) の用量を REBETOL と組み合わせて比較し、Pegasys® (180 mcg/週)/Copegus® (1000-1200 mg/日) を投与された 3 番目の治療群を比較)。
- 研究 5 – 以前の治療失敗の被験者で、体重ベースの REBETOL と組み合わせて PegIntron (1.5 mcg/kg/週) を評価しました。
- 小児患者における PegIntron/REBETOL 200mg 併用療法
- REBETOL/INTRON A 併用療法の成人および小児科向け試験
REBETOL の有無にかかわらず、PegIntron を使用した臨床試験では、被験者の約 12% で重篤な副作用が発生しました [参照 囲み警告 、 警告と注意事項 ]。 PegIntron と REBETOL 200mg で治療された被験者に発生する最も一般的な重篤なイベントは、うつ病と自殺念慮でした [参照 警告と注意事項 ]、それぞれ 1% 未満の頻度で発生します。自殺念慮または自殺企図は、成人患者と比較して、治療中および治療外のフォローアップ中に、小児患者、主に思春期の患者でより頻繁に発生しました(2.4%対1%)。 警告と注意事項 ]。 PegIntron および REBETOL 200mg で治療された被験者に発生する最も一般的な致命的な反応は、心停止、自殺念慮、および自殺未遂でした [参照 警告と注意事項 ]、すべて被験者の 1% 未満で発生します。
臨床試験はさまざまな条件下で実施されるため、ある薬剤の臨床試験で観察された副作用の発生率を別の薬剤の臨床試験で観察された発生率と直接比較することはできず、臨床現場で観察された発生率を反映していない可能性があります。
臨床試験の経験 – REBETOL/PegIntron 併用療法
成人対象
5% を超える発生率で臨床試験で発生した有害反応は、表 5 の REBETOL/PegIntron 併用療法 (研究 2) の治療群によって提供されます。
表 6 は、研究 4 で 10% 以上の発生率で発生した治療関連の有害反応をまとめたものです。
重篤な副作用の発生率は、すべての試験で同等でした。研究 3 では、体重ベースの REBETOL グループ (12%) と固定用量の REBETOL レジメンで同様の重篤な副作用の発生率が報告されました。研究 2 では、重篤な副作用の発生率は、INTRON A/REBETOL グループの 14% と比較して、PegIntron/REBETOL グループで 17% でした。
すべてではありませんが、多くの場合、副作用は減量または治療の中止後に解消されました。一部の被験者は、6か月の追跡期間中に進行中または新たな重篤な副作用を経験しました。研究 2 では、多くの被験者は、治療の中止後数ヶ月にわたって副作用を経験し続けました。 6 か月の追跡期間の終わりまでに、PegIntron 1.5/REBETOL 200mg 群の体格別の進行中の有害反応の発生率は、33% (精神科)、20% (筋骨格系)、および 10% (内分泌および神経系) でした。 GIの場合)。被験者の約 10 ~ 15% で、体重減少、疲労、頭痛が解消されませんでした。
これらの臨床試験では、治療中またはフォローアップ中に 31 人の被験者が死亡しました。研究 1 では、PegIntron 単剤療法を受けた被験者で 1 人の自殺があり、INTRON A 単剤療法を受けた被験者で 2 人の死亡がありました (1 人の殺人/自殺と 1 人の突然死)。研究 2 では、PegIntron/REBETOL 200mg 併用療法を受けた被験者で 1 人の自殺がありました。 INTRON A/REBETOL 200mg 群で 1 例の死亡例(自動車事故)。研究 3 では 14 人が死亡し、そのうち 2 人は自殺の可能性があり、1 人は関連するうつ病の病歴を持つ人の原因不明の死亡でした。研究 4 では、12 人の死亡があり、そのうち 6 人は PegIntron/REBETOL 併用療法を受けた被験者で発生し、5 人は PegIntron 1.5 mcg/REBETOL 群 (N=1019)、1 人は PegIntron 1 mcg/REBETOL 群 (N= 1016)、そのうち 6 件は Pegasys/Copegus を投与された被験者 (N=1035) で発生しました。 PegIntron (1.5 mcg/kg)/REBETOL 200 mg の併用療法を受けた被験者では、非治療のフォローアップ期間中に 3 人の自殺が発生しました。
研究 1 および 2 では、PegIntron 単独または REBETOL 200mg と組み合わせて投与された被験者の 10 ~ 14% が治療を中止しましたが、INTRON A 単独で治療された 6% および REBETOL と組み合わせて INTRON A で治療された 13% と比較されました。同様に、研究 3 では、PegIntron を体重ベースの REBETOL と組み合わせて投与された被験者の 15%、および PegIntron と一定用量の REBETOL 200mg を投与された被験者の 14% が、有害反応のために治療を中止しました。治療中止の最も一般的な理由は、精神医学的、全身的(例えば、疲労、頭痛)、または胃腸の有害反応の既知のインターフェロン効果に関連していました。研究 4 では、PegIntron 1.5 mcg/REBETOL 200mg 群の被験者の 13%、PegIntron 1 mcg/REBETOL 200mg 群の 10%、Pegasys 180 mcg/Copegus 群の 13% が有害事象のために中止されました。
研究 2 では、PegIntron (1.5 mcg/kg)/REBETOL 200mg を投与された被験者の 42%、INTRON A/REBETOL を投与された被験者の 34% で、副作用による用量の減少が発生しました。 PegIntron (1.5 mcg/kg)/REBETOL を投与された体重 60 kg 以下の被験者の大多数 (57%) は、減量を必要としました。インターフェロンの減少は用量依存的であり (PegIntron 1.5 mcg/kg は PegIntron 0.5 mcg/kg または INTRON A よりも大きい)、それぞれ 40%、27%、28% でした。 REBETOL 200mg の減量は、3 つのグループすべてで 33 ~ 35% と同様でした。用量変更の最も一般的な理由は、好中球減少症 (18%) または貧血 (9%) でした ( 検査値 )。その他の一般的な理由には、うつ病、疲労、吐き気、血小板減少症などがあります。研究 3 では、副作用による用量変更は、体重ベースの投薬 (WBD) で、均一投薬よりも頻繁に発生しました (それぞれ 29% と 23%)。研究 4 では、被験者の 16% が REBETOL 200mg と組み合わせて PegIntron を 1 mcg/kg に減量し、さらに 4% が 15% と比較して、有害事象のために PegIntron を 0.5 mcg/kg に 2 回減量する必要がありました。ペガシス/コペガス群の被験者はペガシスで135 mcg/週への減量を必要とし、ペガシス/コペガス群ではさらに7%がペガシスで90 mcg/週への2回目の減量を必要としました.
PegIntron/REBETOL 200mg 併用試験では、最も一般的な副作用は精神疾患であり、研究 2 では被験者の 77%、研究 3 では被験者の 68% ~ 69% で発生しました。不眠症は、すべての治療グループの被験者の約 30% から 40% によってそれぞれ報告されました。治療中または治療中止後のフォローアップ中に、全被験者の 2% で自殺行動 (観念、自殺未遂、および自殺) が発生した [参照 警告と注意事項 ]。研究 4 では、PegIntron 1.5 mcg/REBETOL 200mg 群の被験者の 58%、PegIntron 1 mcg/REBETOL 200mg 群の被験者の 55%、Pegasys 180 mcg/Copegus 群の被験者の 57% で精神医学的副作用が発生しました。 .
PegIntron は被験者の約 3 分の 2 に疲労または頭痛を誘発し、被験者の約半数に発熱または悪寒を引き起こしました。これらの全身症状の一部(発熱や頭痛など)の重症度は、治療を続けるにつれて軽減する傾向がありました。研究 1 および 2 では、適用部位の炎症および反応 (例えば、打撲、かゆみ、刺激) は、INTRON A と比較して、PegIntron 療法の約 2 倍の発生率 (被験者の最大 75%) で発生しました。すべてのグループでまれ (2 ~ 3%)。研究 3 では、注射部位反応または炎症の発生率は全体で 23% から 24% でした。
以前のインターフェロン併用レジメンで失敗した後、再治療として REBETOL/PegIntron を投与された被験者は、未治療の被験者の臨床試験中に、以前にこのレジメンに関連したものと同様の副作用を報告しました。
小児科
一般に、小児集団における有害反応プロファイルは、成人で観察されたものと同様でした。小児科の試験では、すべての被験者で最も一般的な副作用は、発熱 (80%)、頭痛 (62%)、好中球減少 (33%)、疲労 (30%)、食欲不振 (29%)、注射部位紅斑 (29%) でした。 %) と嘔吐 (27%)。試験で報告された副作用の大部分は、軽度または中程度の重症度でした。重度の有害反応は、すべての被験者の 7% (8/107) で報告され、注射部位の痛み (1%)、四肢の痛み (1%)、頭痛 (1%)、好中球減少症 (1%)、および発熱 (4%) が含まれていました。 %)。この被験者集団で発生した重要な副作用は、神経質 (7%; 7/107)、攻撃性 (3%; 3/107)、怒り (2%; 2/107)、および抑うつ (1%; 1/107) でした。 . 5 人の被験者がレボチロキシン治療を受け、そのうち 3 人は臨床的甲状腺機能低下症、2 人は無症候性の TSH 上昇でした。 PegIntron と REBETOL で治療された小児被験者の体重と身長の増加は、治療の全期間にわたって標準集団データによって予測されたものよりも遅れていました。治療中の被験者の70%で、重度の成長速度の阻害(3パーセンタイル未満)が観察されました。
PegIntron および/またはリバビリンの用量変更は、貧血、好中球減少症、および体重減少が最も一般的である治療関連の有害反応のために、被験者の 25% で必要とされました。 2 人の被験者 (2%; 2/107) は、副作用の結果として治療を中止しました。
小児科の被験者で 10% 以上の発生率で発生した有害反応を表 7 に示します。
107 人の被験者のうち 94 人が 5 年間の長期追跡試験に登録されました。成長に対する長期的な影響は、48 週間治療した被験者よりも 24 週間治療した被験者の方が少なかった. 24 週間治療を受けた被験者の 24% (11/46) および 48 週間治療された被験者の 40% (19/48) は、治療前から 5 年の終わりまでの年齢に対する身長の減少が 15 パーセンタイルを超えていました。治療前のベースラインパーセンタイルと比較した長期追跡。 24 週間治療を受けた被験者の 11% (5/46)、および 48 週間治療された被験者の 13% (6/48) で、治療前のベースラインから年齢に対する身長パーセンタイルが 30 を超えて減少したことが観察されました。 5年間の長期追跡調査。すべての年齢層で観察されましたが、長期追跡調査の終了時に身長が減少するリスクが最も高いのは、予想される最大成長速度の年に併用療法を開始した場合と相関しているように見えました。 [見る 警告と注意事項 ]
検査値
成人および小児科
PegIntron/体重ベースの REBETOL 200mg 併用療法と PegIntron/一定用量の REBETOL 200mg レジメンを比較した試験 3 の有害反応プロファイルでは、体重ベースの投薬で貧血の割合が増加することが明らかになりました (体重ベースの投薬では 29% 対 19%)。対フラット用量レジメン、それぞれ)。しかし、貧血の症例の大部分は軽度であり、用量の減少に反応しました。
レベトール 200mg 治療と組み合わせた治療中の選択された臨床検査値の変化を以下に示します。 ヘモグロビン、白血球、好中球、血小板の減少により、用量を減らすか、治療を永久に中止する必要がある場合があります [見る 投薬と管理 ]。治療中の選択された検査値の変化を表 8 に示します。小児科を対象とした PegIntron/REBETOL 200mg 試験における検査値の変化のほとんどは、軽度または中等度でした。
ヘモグロビン
研究 2 では、被験者の約 30% でヘモグロビン値が 11 g/dL 未満に減少しました。 /dl。 9 g/dL 未満へのヘモグロビンの減少は、均一投与と比較して WBD を受けた被験者でより頻繁に発生しました (それぞれ 4% および 2%)。試験 2 では、PegIntron/REBETOL 200mg および INTRON A/REBETOL 200mg 群の被験者の 9% および 13% で用量変更が必要でした。研究 4 では、PegIntron (1.5 mcg/kg)/REBETOL を投与された被験者は、ヘモグロビン レベルが 8.5 から 10 g/dL 未満 (28%) および 8.5 g/dL 未満 (3%) に減少しました。 Pegasys 180 mcg/Copegus を投与された場合、これらの減少はそれぞれ被験者の 26% と 4% で発生しました。ヘモグロビンレベルは、平均して4~6週間の治療で安定しました。観察された典型的なパターンは、4週目の治療によるヘモグロビンレベルの低下、その後の安定化とプラトーであり、これは治療の終わりまで維持されました。 PegIntron 単剤療法の試験では、ヘモグロビンの減少は一般的に軽度であり、用量の変更が必要になることはめったにありませんでした。 投薬と管理 ]。
好中球
好中球数の減少は、研究 2 の REBETOL 200mg (85%) および INTRON A/REBETOL (60%) による併用療法で治療された成人被験者の大部分で観察されました。生命を脅かす可能性のある重度の好中球減少症 (0.5 x 109/L 未満) は、INTRON A/REBETOL 200mg で治療された被験者の 2%、および試験 2 で PegIntron/REBETOL 200mg で治療された被験者の約 4% で発生しました。スタディ 2 の PegIntron/REBETOL では、インターフェロン投与量の変更が必要でした。少数の被験者 (1% 未満) は、治療を永久に中止する必要がありました。好中球数は一般に、治療中止後 4 週間で治療前のレベルに戻った [参照 投薬と管理 ]。
血小板
血小板数は、PegIntron 単独または REBETOL 200mg で治療された被験者の約 20%、および INTRON A/REBETOL で治療された成人被験者の 6% で 100,000/mm³ 未満に減少しました。血小板数の深刻な減少 (50,000/mm³ 未満) は、成人被験者の 4% 未満で発生します。血小板減少の結果として、患者は中止または用量変更を必要とする場合があります [参照 投薬と管理 ]。研究 2 では、被験者の 1% または 3% がそれぞれ INTRON A または PegIntron の用量変更を必要としました。血小板数は一般に、治療中止後 4 週間で治療前のレベルに戻りました。
甲状腺機能
TSH 異常の発生は、臨床症状の有無にかかわらず、インターフェロン療法に関連しています。研究 2 では、INTRON A または PegIntron (REBETOL の有無にかかわらず) で治療された被験者の間で、臨床的に明らかな甲状腺障害が同様の発生率 (甲状腺機能低下症で 5%、甲状腺機能亢進症で 3%) で発生しました。被験者は、治療中および追跡期間中に新たな TSH 異常を発症しました。追跡調査期間の終わりに、被験者の 7% はまだ異常な TSH 値を持っていました。
ビリルビンと尿酸
研究 2 では、被験者の 10 ~ 14% が高ビリルビン血症を発症し、33 ~ 38% が溶血に伴う高尿酸血症を発症しました。 6人の被験者が軽度から中等度の痛風を発症しました。
治験経験 – レベトール/イントロンA併用療法
成人対象
臨床試験では、インターフェロン群の 13% と 3% と比較して、以前に治療を受けていない被験者と再発した被験者のそれぞれ 19% と 6% が、併用群で副作用のために治療を中止しました。 5%以上の発生率で米国の試験で発生した選択された治療関連の有害反応は、治療グループによって提供されます (表 9 を参照)。一般に、無力症、インフルエンザ様症状、神経過敏、および掻痒を除いて、選択された治療関連の有害反応は、米国の試験と比較して国際試験でより低い発生率で報告されました。
小児科
歳から 16 歳までの 118 人の小児被験者を対象とした臨床試験では、6% が副作用のために治療を中止しました。被験者の 30% で用量変更が必要であり、最も一般的なのは貧血と好中球減少症です。一般に、小児集団における有害反応プロファイルは、成人で観察されたものと同様でした。注射部位障害、発熱、食欲不振、嘔吐、および情緒不安定は、成人被験者と比較して小児被験者でより頻繁に発生しました。逆に、小児被験者は、成人被験者と比較して、疲労、消化不良、関節痛、不眠症、過敏性、集中力の低下、呼吸困難、およびそう痒症を経験しませんでした。 REBETOL/INTRON A 併用療法の推奨用量を受けたすべての小児被験者の間で 5% 以上の発生率で発生した選択された治療関連の有害反応を表 9 に示します。
48 週間の治療過程で、直線的な成長率の低下 (パーセンタイル割り当ての平均 7% の減少) と体重増加率の減少 (パーセンタイル割り当ての平均 9% の減少) が見られました。これらの傾向の一般的な逆転は、治療後 24 週間で認められました。しかし、限られた数の患者における長期データは、併用療法が一部の患者で最終的な成人の身長の低下をもたらす成長阻害を誘発する可能性があることを示唆している[参照 警告と注意事項 ]。
検査値
治療中の選択された血液学的値 (ヘモグロビン、白血球、好中球、および血小板) の変化を以下に示します (表 10 を参照)。
ヘモグロビン。 REBETOL 療法を受けた被験者のヘモグロビンの減少は 1 週目に始まり、4 週目までに安定しました。48 週間治療された未治療の被験者では、ベースラインからの平均最大減少量は、米国の試験で 3.1 g/dL、国際試験で 2.9 g/dL でした。トライアル。再発被験者では、ベースラインからの平均最大減少量は、米国の試験で 2.8 g/dL、国際試験で 2.6 g/dL でした。ヘモグロビン値は、ほとんどの被験者で、治療中止後 4 ~ 8 週間以内に治療前のレベルに戻りました。
ビリルビンと尿酸. 臨床試験では、溶血に関連するビリルビンと尿酸の両方の増加が認められました。ほとんどは中等度の生化学的変化であり、治療中止後 4 週間以内に元に戻りました。この観察は、以前にギルバート症候群と診断された被験者で最も頻繁に発生しました。これは、肝機能障害や臨床的罹患率とは関連していません。
市販後の経験
REBETOL と INTRON A または PegIntron を併用した承認後の使用中に、以下の有害反応が特定され、報告されています。これらの反応は不確かな規模の集団から自発的に報告されるため、その頻度を確実に推定したり、薬物曝露との因果関係を確立したりすることは常に可能ではありません.
血液およびリンパ系の疾患
赤芽球癆、再生不良性貧血
耳と迷路の障害
聴覚障害、めまい
呼吸器、胸部および縦隔の障害
肺高血圧症
眼疾患
漿液性網膜剥離
内分泌疾患
糖尿病
薬物相互作用
ジダノシン
ジダノシンまたはその活性代謝物 (ジデオキシアデノシン 5'-三リン酸) への曝露は、ジダノシンをリバビリンと併用すると増加し、臨床毒性を引き起こしたり悪化させたりする可能性があります。したがって、レベトール 200mg カプセルまたは内服液とジダノシンの併用は禁忌です。致命的な肝不全、末梢神経障害、膵炎、症候性高乳酸血症/乳酸アシドーシスの報告が臨床試験で報告されています。
ヌクレオシド類似体
肝臓の代償不全(致死的なものもある)は、HIV とインターフェロン アルファおよびリバビリンの併用抗レトロ ウイルス療法を受けている肝硬変 HIV/HCV 同時感染患者で発生しています。アルファインターフェロン単独またはリバビリンとの併用による治療を追加すると、この患者集団のリスクが高まる可能性があります。リバビリンおよびヌクレオシド系逆転写酵素阻害剤 (NRTI) とともにインターフェロンを投与されている患者は、治療に関連する毒性、特に肝代償不全および貧血について注意深く監視する必要があります。 NRTI の中止は、医学的に適切であると考えるべきです( 個々の NRTI 製品のラベル付け )。インターフェロン、リバビリン、またはその両方の減量または中止も、肝代償不全などの臨床毒性の悪化が観察された場合(例、Child-Pughが6を超える場合)を考慮する必要があります。
リバビリンは、HIV に対するスタブジンおよびジドブジンの細胞培養抗ウイルス活性に拮抗する可能性があります。リバビリンは細胞培養において、ラミブジン、スタブジン、およびジドブジンのリン酸化を阻害することが示されており、抗レトロウイルス活性の低下につながる可能性があります。しかし、別のペグ化インターフェロンとリバビリンを併用した研究では、リバビリンとラミブジンを併用した場合、薬物動態学的相互作用 (例、血漿濃度または細胞内三リン酸化活性代謝物濃度) または薬力学的相互作用 (例、HIV/HCV ウイルス抑制の喪失) は観察されませんでした (n =18)、スタブジン (n=10)、またはジドブジン (n=6) が、HIV/HCV 同時感染被験者の多剤レジメンの一部として同時投与されました。したがって、リバビリンとこれらの薬剤の併用には注意が必要です。
シトクロム P-450 によって代謝される薬物
ヒトおよびラットの両方の肝臓ミクロソーム調製物を使用した in vitro 試験の結果は、シトクロム P-450 酵素を介したリバビリンの代謝がほとんどまたはまったくないことを示しており、P-450 酵素に基づく薬物相互作用の可能性は最小限でした。
複数回投与の薬物動態研究において、INTRON A と REBETOL 200mg カプセルの間に薬物動態学的相互作用は認められませんでした。
アザチオプリン
アザチオプリンを投与されている慢性 C 型肝炎患者の治療にリバビリンを使用すると、重度の汎血球減少症が誘発され、アザチオプリン関連の骨髄毒性のリスクが高まる可能性があることが報告されています。イノシン一リン酸デヒドロゲナーゼ (IMDH) は、アザチオプリンの代謝経路の 1 つに必要です。リバビリンは IMDH を阻害することが知られているため、骨髄毒性 (好中球減少症、血小板減少症、および貧血) に関連するアザチオプリン代謝産物である 6-メチルチオイノシン一リン酸 (6-MTITP) が蓄積されます。アザチオプリンとリバビリンの併用療法を受けている患者は、血小板数を含む完全な血球計算を、治療の最初の 1 か月間は毎週、2 か月目と 3 か月目は 1 か月に 2 回、その後は 1 か月に 1 回、または投与量や他の治療法の変更が必要な場合はより頻繁に行う必要があります。 警告と注意事項 ]。
警告
の一部として含まれています 予防 セクション。
予防
妊娠
REBETOL 200mg カプセルおよび経口溶液は、先天性欠損症および胎児の死亡を引き起こす可能性があります。 REBETOL 療法は、計画された療法開始の直前に妊娠検査で陰性の報告が得られるまで開始しないでください。患者は少なくとも 2 種類の避妊法を使用し、治療中および治療中止後 6 か月間は毎月妊娠検査を受ける必要があります。 女性患者および男性患者の女性パートナーの妊娠を避けるために、細心の注意を払う必要があります。 REBETOL 200mg が実証済み 適切な研究が実施されたすべての動物種における重大な催奇形性および殺胚性効果。これらの影響は、次の用量で発生しました。 リバビリンの推奨ヒト用量の 20 分の 1 という低用量です。 REBETOL 200mg の治療は、妊娠検査で陰性と報告されるまで開始しないでください。 計画された治療開始の直前に取得されている[参照 囲み警告 、 禁忌 、 特定の集団での使用 、 と 患者情報 ]。
貧血
リバビリンの主な毒性は溶血性貧血であり、これは臨床試験でレベトール/イントロン A で治療された被験者の約 10% で観察されました。レベトール カプセルに伴う貧血は、治療開始から 1 ~ 2 週間以内に発生します。ヘモグロビンの初期低下が著しい場合があるため、ヘモグロビンまたはヘマトクリットを治療開始前および治療の 2 週目と 4 週目に、または臨床的に必要な場合はより頻繁に測定することをお勧めします。その後、患者は臨床的に適切な経過をたどる必要があります[参照 投薬と管理 ]。
REBETOL による貧血患者では、致命的および非致命的な心筋梗塞が報告されています。リバビリン療法を開始する前に、基礎にある心疾患について患者を評価する必要があります。既存の心疾患のある患者は、治療前に心電図を投与する必要があり、治療中は適切に監視する必要があります。心血管状態の悪化があれば、治療を中断または中止する必要があります[参照 投薬と管理 ]。心疾患は薬剤性貧血によって悪化する可能性があるため、重大または不安定な心疾患の既往歴のある患者は、REBETOL を使用しないでください。
膵炎
REBETOL と INTRON A または PegIntron による治療は、膵炎の兆候と症状がある患者では中止し、膵炎が確認された患者では中止する必要があります。
肺疾患
呼吸困難、肺浸潤、肺臓炎、肺高血圧症、および肺炎を含む肺の症状が、αインターフェロン併用療法を伴うREBETOLによる治療中に報告されています。致命的な肺炎の時折のケースが発生しました。また、サルコイドーシスやサルコイドーシスの増悪が報告されています。肺浸潤または肺機能障害の証拠がある場合、患者を綿密に監視し、適切であれば併用療法を中止する必要があります。
眼科疾患
リバビリンは、アルファ インターフェロンとの併用療法で使用されます。視力の低下または喪失、黄斑浮腫、網膜動脈または静脈を含む網膜症、血栓症、網膜出血および脱脂綿斑点、視神経炎、乳頭浮腫、および漿液性網膜剥離が、アルファインターフェロンによる治療によって誘発または悪化します。すべての患者は、ベースラインで目の検査を受ける必要があります。既存の眼科疾患(糖尿病または高血圧性網膜症など)のある患者は、アルファインターフェロン治療との併用療法中に定期的な眼科検査を受けるべきです。眼症状を発症した患者は、迅速かつ完全な眼科検査を受ける必要があります。 αインターフェロンとの併用療法は、眼科疾患が新たに発生したり悪化したりした患者では中止する必要があります。
臨床検査
PegIntron をリバビリンと併用すると、好中球数と血小板数が大幅に減少し、血液、内分泌(TSH など)、および肝臓の異常が生じる可能性があります。
PegIntron/REBETOL 併用療法を受けている患者は、治療開始前に血液学および血液化学検査を受け、その後も定期的に検査を受ける必要があります。成人臨床試験では、2、4、8、12 週、およびその後は 6 週間間隔で、または異常が発生した場合はより頻繁に。小児被験者では、治療 6 週目にヘモグロビンを追加評価して、同じ検査パラメータを評価しました。治療期間中、TSH レベルを 12 週間ごとに測定しました。 HCV-RNA は、治療中に定期的に測定する必要があります [ 投薬と管理 ]。
歯と歯周病
リバビリンとインターフェロンまたはペグインターフェロンの併用療法を受けている患者では、歯科および歯周疾患が報告されています。さらに、REBETOL とペグ化または非ペグ化インターフェロン α-2b の組み合わせによる長期治療中に、口腔乾燥が歯および口の粘膜に損傷を与える可能性があります。患者は 1 日 2 回徹底的に歯を磨き、定期的に歯科検診を受ける必要があります。嘔吐が発生した場合は、その後、口を完全にすすぐようにアドバイスする必要があります。
アザチオプリンの同時投与
ペグ化インターフェロン/リバビリンとアザチオプリンの同時投与後、3~7週間以内に汎血球減少症(赤血球、好中球、および血小板の顕著な減少)および骨髄抑制が起こることが文献で報告されています。この限られた数の患者 (n=8) では、骨髄毒性は、HCV 抗ウイルス療法と併用アザチオプリンの両方を中止すると 4 ~ 6 週間以内に回復し、いずれかの治療のみを再開しても再発しませんでした。 PegIntron、REBETOL、およびアザチオプリンは、汎血球減少症のために中止する必要があり、ペグ化インターフェロン/リバビリンは、併用アザチオプリンと一緒に再導入すべきではありません[ 薬物相互作用 ]。
成長への影響 - 小児への使用
成長に対する PegIntron と REBETOL の効果に関するデータは、3 歳から 17 歳までの被験者を対象とした非盲検研究から得られたもので、体重と身長の変化が米国の標準人口データと比較されています。一般に、PegIntron および REBETOL 200mg で治療された小児被験者の体重および身長の増加は、治療期間全体の標準集団データによって予測されたものよりも遅れています。治療中の被験者の70%で、重度の成長速度の阻害(3パーセンタイル未満)が観察されました。治療後、ほとんどの被験者でリバウンド成長と体重増加が起こりました。しかし、小児被験者の長期追跡データは、REBETOL との併用療法における PegIntron が、一部の患者で成人の身長の低下をもたらす成長阻害を誘発する可能性があることを示しています [ 有害反応 ]。
同様に、レベトール 200mg とイントロン A の併用療法を 1 年間行った後、被験者の成長に影響が見られました。これらの被験者の限られた数の長期追跡試験では、併用療法により、一部の被験者の最終的な成人の身長が減少しました[参照 有害反応 ]。
使用上の保護
臨床試験の結果に基づくと、リバビリン単剤療法は慢性 C 型肝炎ウイルス感染症の治療には有効ではありません。したがって、レベトール 200mg カプセルまたは経口溶液を単独で使用してはなりません。 REBETOL カプセルおよび経口溶液の安全性と有効性は、INTRON A または PegIntron (他のインターフェロンではありません) と組み合わせて使用した場合にのみ確立されています。
HIV 感染症、アデノウイルス、RSV、パラインフルエンザ、またはインフルエンザ感染症の治療における REBETOL/INTRON A および PegIntron 療法の安全性と有効性は確立されていません。レベトール 200mg カプセルは、これらの適応症には使用しないでください。吸入用リバビリンには別のラベルがあり、リバビリン吸入療法を検討している場合は参照する必要があります。
REBETOL/INTRON A または PegIntron 療法によって引き起こされる重大な副作用があり、重度のうつ病および自殺念慮、溶血性貧血、骨髄機能の抑制、自己免疫疾患および感染症、肺機能障害、膵炎、および糖尿病が含まれます。自殺念慮または自殺企図は、治療中および治療外のフォローアップ中に、成人患者と比較して、主に青年期の小児患者でより頻繁に発生しました (2.4% 対 1%)。 INTRON A および PegIntron のラベル表示は、併用療法を開始する前に追加の安全性情報を得るために全体を確認する必要があります。
患者相談情報
患者に FDA 承認の患者ラベリング ( 投薬ガイド )。
貧血
レベトール カプセルで発生する最も一般的な有害事象は貧血であり、重篤な場合があります [参照 警告と注意事項 と 有害反応 ]。患者は、治療開始前とその後定期的に検査室での評価が必要であることを知らされるべきである[参照 投薬と管理 ]。特に治療の初期段階では、十分に水分を補給することをお勧めします。
妊娠
患者は、REBETOL 200mg カプセルおよび経口溶液が先天異常および胎児の死亡を引き起こす可能性があることを知らされなければなりません。 REBETOL 200mg は、妊娠中の女性、または女性パートナーが妊娠している男性は使用しないでください。女性患者およびレベトールを服用している男性患者の女性パートナーの妊娠を避けるために、細心の注意を払う必要があります。 REBETOL は、治療開始直前に妊娠検査で陰性の報告が得られるまで開始しないでください。患者は、治療中および治療後 6 か月間、毎月妊娠検査を実施する必要があります。
出産の可能性のある女性は、治療を開始する前に、効果的な避妊法(信頼できる 2 つの方法)の使用についてカウンセリングを受ける必要があります。患者(男性および女性)は、催奇形性/殺胚性リスクについて知らされ、REBETOL 200mg の投与中および治療後 6 か月間、効果的な避妊を実践するように指示されなければなりません。患者(男性および女性)は、妊娠の場合には直ちに医師に通知するように助言されるべきである[参照 禁忌 、 警告と注意事項 、 と 特定の集団での使用 ]。
治療中または治療後 6 か月の間に妊娠が発生した場合、患者は、胎児に対する REBETOL 200mg 治療の催奇形性リスクについて知らされなければなりません。患者または患者のパートナーは、治療中または治療中止後 6 か月以内に妊娠が発生した場合は、直ちに主治医に報告する必要があります。処方者は、1-800-593-2214 に電話して、そのような場合を報告する必要があります。
リスクとメリット
REBETOL カプセルを服用している患者は、治療に関連する利点とリスクを知らされ、適切な使用方法を指示され、患者に紹介されるべきです。 投薬ガイド . C 型肝炎感染症の治療が感染に及ぼす影響は不明であること、および C 型肝炎ウイルスの感染を防ぐための適切な予防措置を講じる必要があることを患者に知らせる必要があります。
患者は、REBETOL の投与を逃した場合に何をすべきかについて知らされるべきです。逃した用量は、同じ日にできるだけ早く服用する必要があります。患者は次の用量を 2 倍にしないでください。質問がある場合は、医療提供者に連絡するよう患者に助言する必要があります。
非臨床毒性学
発がん、突然変異誘発、生殖能力の障害
発がん
リバビリンは、トランスジェニック p53 欠損マウスモデルに最大 300 mg/kg の用量で 6 か月間投与した場合、どの腫瘍タイプにも増加を引き起こしませんでした (60 kg の成人の体表面積調整に基づく推定ヒト等価量は 25 mg/kg です)。 ; ヒトの 1 日最大推奨用量の約 1.9 倍)。リバビリンは、最大 40 mg/kg の用量でラットに 2 年間投与された場合、発がん性がありませんでした (60 kg の成人の体表面積調整に基づくヒトの推定値は 5.71 mg/kg です)。
突然変異誘発
リバビリンは、複数の遺伝毒性アッセイで突然変異と細胞形質転換の発生率の増加を示しました。リバビリンは、Balb/3T3 In Vitro 細胞形質転換アッセイで活性でした。変異原性活性は、マウスのリンパ腫アッセイで観察され、20 ~ 200 mg/kg の用量で観察されました (60 kg の成人の体表面積調整に基づいて、1.67 ~ 16.7 mg/kg の推定ヒト等価量; 最大値の 0.1 ~ 1 倍)。マウス小核アッセイにおけるリバビリンの推奨ヒト 24 時間用量)。ラットの優性致死アッセイは陰性であり、ラットに突然変異が発生した場合、それらは雄の配偶子を介して伝達されなかったことを示しています.
受胎能障害
リバビリンは、適切な研究が実施されたすべての動物種において、ヒトの推奨用量をはるかに下回る用量で、有意な殺胚および催奇形効果を示しました。頭蓋骨、口蓋、目、顎、手足、骨格、消化管の奇形が認められました。催奇形性の影響の発生率と重症度は、薬剤の用量が増加するにつれて増加しました。胎児と子孫の生存率が低下しました。ラットとウサギでの従来の胚毒性/催奇形性研究では、観察された無影響用量レベルは、提案された臨床使用の用量レベルをはるかに下回っていました (ラットとウサギの両方で 0.3 mg/kg/日; 推奨されるヒトの 24 時間用量の約 0.06 倍)。リバビリン)。 1 mg/kg/日まで経口投与されたラットの周産期/出生後毒性試験で、母体毒性または子孫への影響は観察されませんでした (60 kg の成人の体表面積調整に基づく推定ヒト等価用量は 0.17 mg/kg です)。 ; リバビリンのヒト 24 時間最大推奨用量の約 0.01 倍) [参照 禁忌 、 と 警告と注意事項 ]。
妊娠可能な女性とそのパートナーは、患者とそのパートナーが効果的な避妊法 (2 つの信頼できる方法) を使用していない限り、REBETOL を受けるべきではありません。リバビリンの反復投与半減期 (t1/2) が 12 日間であることから、治療後 6 か月間は有効な避妊法を使用する必要があります (例えば、リバビリンのクリアランスの半減期は 15 回)。
REBETOL は、妊娠可能な男性には注意して使用する必要があります。 15 ~ 150 mg/kg/日の用量でのリバビリン誘発性精巣変性の経時変化と可逆性を評価するためのマウスでの研究では、 60 kg の成人; リバビリンの最大ヒト 24 時間用量の 0.1 ~ 0.8 倍)を 3 ~ 6 か月間投与すると、精子に異常が発生しました。治療を中止すると、リバビリン誘発性精巣毒性からの本質的に完全な回復が、1または2回の精子形成周期内で明らかになった。
特定の集団での使用
妊娠
妊娠カテゴリーX
[見る 禁忌 、 警告と注意事項 、 と 非臨床毒性学 ]。
治療と後処理:
胎児への潜在的なリスク
リバビリンは、細胞内成分に蓄積することが知られており、そこから非常にゆっくりと除去されます。精子に含まれるリバビリンが卵子の受精に潜在的な催奇形効果を発揮するかどうかはわかっていません。ラットでの研究では、リバビリンを 200 mg/kg までの用量で 5 日間投与しても優性致死は誘発されなかったと結論付けられました (人間の推定等価用量は 7.14 ~ 28.6 mg/kg、体表面積を 60 mm に調整した場合)。 kg 成人; リバビリンの最大推奨ヒト用量の 1.7 倍まで)。しかし、リバビリンの潜在的なヒト催奇形作用のため、男性患者は、女性パートナーの妊娠のリスクを回避するためにあらゆる予防策を講じるようにアドバイスされるべきです.
出産の可能性のある女性は、治療期間中に効果的な避妊法(信頼できる 2 つの方法)を使用していない限り、REBETOL を受けるべきではありません。さらに、リバビリンの反復投与半減期 (t1/2) が 12 日であることから、治療後 6 か月間は効果的な避妊を行う必要があります。
男性患者とその女性パートナーは、REBETOL による治療中および治療後 6 か月間 (例えば、体からリバビリンが除去される半減期の 15 倍) の間、効果的な避妊 (信頼できる 2 つの形態) を実践しなければなりません。
リバビリン妊娠登録は、治療中および治療中止後 6 か月間、リバビリンにさらされた女性患者および男性患者の女性パートナーの妊娠の母体胎児転帰を監視するために確立されました。医師と患者は、1-800-593-2214 に電話してそのような症例を報告することをお勧めします。
授乳中の母親
レベトール製品が母乳中に排泄されるかどうかは不明です。授乳中の乳児に深刻な副作用が生じる可能性があるため、授乳を中止するか、REBETOL を延期または中止するかを決定する必要があります。
小児用
PegIntron と組み合わせた REBETOL 200mg の安全性と有効性は、3 歳未満の小児患者では確立されていません。 REBETOL/INTRON A による治療では、小児患者の治療を決定する際に、肝炎症や線維症などの疾患進行の証拠、および反応の予後因子、HCV 遺伝子型、およびウイルス量を考慮する必要があります。治療の利点は、観察された安全性の調査結果と比較検討する必要があります。
小児被験者の長期追跡データは、PegIntron または INTRON A と組み合わせた REBETOL が一部の患者で身長の減少をもたらす成長阻害を誘発する可能性があることを示しています [ 警告と注意事項 と 有害反応 ]。
自殺念慮または自殺企図は、成人患者と比較して、治療中および治療外のフォローアップ中に、主に青年期の小児患者でより頻繁に発生しました(2.4%対1%)。 [見る 警告と注意事項 ]。成人患者と同様に、小児患者は他の経験をしました。 精神医学的副作用(例、抑うつ、情緒不安定、傾眠)、貧血、および好中球減少症[参照 警告と注意事項 ]。
高齢者の使用
REBETOL/INTRON A または PegIntron 療法の臨床試験には、65 歳以上の対象者が若い対象者と異なる反応を示すかどうかを判断するのに十分な数の対象者が含まれていませんでした。
レベトール 200mg は実質的に腎臓から排泄されることが知られており、腎機能障害のある患者では、この薬剤に対する毒性反応のリスクが高くなる可能性があります。高齢者は腎機能が低下している場合が多いため、用量選択には注意が必要です。腎機能を監視し、それに応じて投与量を調整する必要があります。 REBETOL 200mg は、クレアチニンクリアランスが 50 mL/min 未満の患者には使用しないでください [ 禁忌 ]。
一般に、レベトール 200mg カプセルは、肝機能および心機能の低下、および付随する疾患または他の薬物療法の頻度が高いことを反映して、投与範囲の下限から開始して、高齢の患者に慎重に投与する必要があります。臨床試験では、年配の被験者は、若い患者 (28%) よりも貧血の頻度が高かった (67%) [ 警告と注意事項 ]。
臓器移植レシピエント
肝臓または他の臓器移植レシピエントにおける C 型肝炎の治療における INTRON A および PegIntron 単独または REBETOL との併用の安全性と有効性は確立されていません。小規模(n=16)の単一施設の制御されていない症例経験では、インターフェロンアルファとリバビリンの併用療法を受けている同種腎移植レシピエントの腎不全は、併用療法を受けていない同種腎移植レシピエントに関するセンターの以前の経験から予想されるよりも頻繁でした。腎不全と腎同種移植片拒絶反応との関係は明らかではありません。
HIVまたはHBVの同時感染
HIV または HBV に同時感染した HCV 患者の治療における PegIntron/REBETOL 200mg および INTRON A/REBETOL の安全性と有効性は確立されていません。
過剰摂取
過剰摂取の経験は限られています。最大 20 g のレベトール カプセルの急性摂取、最大 1 億 2000 万単位のイントロン A 摂取、推奨用量の最大 10 倍のイントロン A の皮下投与が報告されています。観察された主な影響は、INTRON A と REBETOL の治療的使用に関連する副作用の発生率と重症度の増加です。しかし、肝酵素異常、腎不全、出血、および心筋梗塞が、推奨用量を超えるイントロン A の単回皮下投与で報告されています。
INTRON AまたはREBETOL 200mgの過剰摂取に対する特定の解毒剤はなく、血液透析および腹膜透析はこれらの薬剤の過剰摂取の治療には効果的ではありません.
禁忌
REBETOL 併用療法は、次の場合に禁忌です。
- 妊娠中の女性。レベトール 200mg を妊婦に投与すると、胎児に害を及ぼす可能性があります。 REBETOL は、妊娠中または妊娠する可能性のある女性には禁忌です。妊娠中に REBETOL を使用する場合、または患者が REBETOL 200mg を服用中に妊娠した場合、患者は胎児への潜在的な危険性について知らされるべきです [ 警告と注意事項 、 特定の集団での使用 、 と 患者情報 ]
- 女性パートナーが妊娠している男性
- スティーブンス・ジョンソン症候群、中毒性、表皮壊死融解症、リバビリンまたは製品の成分に対する多形紅斑などの既知の過敏反応のある患者
- 自己免疫性肝炎患者
- 異常ヘモグロビン症(例,サラセミア・メジャー,鎌状赤血球貧血)の患者
- クレアチニンクリアランスが 50 mL/min 未満の患者。 [見る 特定の集団での使用 と 臨床薬理学 ]
- REBETOL とジダノシンの同時投与は、ジダノシンの活性代謝物 (ジデオキシアデノシン 5'-三リン酸) への曝露が増加するため、禁忌です。ジダノシンとリバビリンの併用療法を受けている患者では、致命的な肝不全、末梢神経障害、膵炎、症候性高乳酸血症/乳酸アシドーシスが報告されている[参照 薬物相互作用 ]。
臨床薬理学
作用機序
リバビリンは抗ウイルス剤です [ 微生物学 ]。
薬物動態
成人における単回および複数回投与の薬物動態特性を表 11 にまとめます。リバビリンは、経口投与後に迅速かつ広範囲に吸収されました。ただし、初回通過代謝のため、絶対バイオアベイラビリティは平均 64% (44%) でした。リバビリン 200 ~ 1200 mg の単回投与後、投与量と AUCtf (時間ゼロから最後の測定可能な濃度までの AUC) の間には直線関係がありました。用量と Cmax の関係は曲線であり、400 ~ 600 mg の単回用量を超えると漸近する傾向がありました。
反復経口投与により、AUC12hr に基づいて、リバビリンの 6 倍の蓄積が血漿中に観察されました。 600 mg を 1 日 2 回経口投与した後、約 4 週間で定常状態に達し、平均定常状態血漿濃度は 2200 ng/mL (37%) でした。投与中止時の平均半減期は 298 (30%) 時間であり、これはおそらく非血漿コンパートメントからのゆっくりとした排出を反映しています。
リバビリンの吸収に対する制酸剤の効果
マグネシウム、アルミニウム、およびシメチコンを含む制酸剤とレベトール カプセルを同時投与すると、平均リバビリン AUCtf が 14% 減少しました。この単回投与試験の結果の臨床的関連性は不明です。
組織分布
非血漿コンパートメントへのリバビリンの輸送は、赤血球で最も広く研究されており、主に es 型の平衡ヌクレオシド輸送体を介して行われることが確認されています。このタイプのトランスポーターは、事実上すべての細胞タイプに存在し、大量の分布の原因となる可能性があります。リバビリンは血漿タンパク質に結合しません。
代謝と排泄
リバビリンには 2 つの代謝経路があります。(i) 有核細胞における可逆的リン酸化経路。 (ii) 脱リボシル化およびアミド加水分解を含む分解経路により、トリアゾールカルボン酸代謝物が生成されます。リバビリンとそのトリアゾールカルボキサミドおよびトリアゾールカルボン酸代謝物は腎臓から排泄されます。 14C-リバビリン 600 mg を経口投与した場合、336 時間で尿中に約 61%、糞中に約 12%の放射能が排泄された。変更されていないリバビリンは、投与量の 17% を占めていました。
特別な集団
腎機能障害
リバビリンの薬物動態は、リバビリンの単回経口用量 (400 mg) を、さまざまな程度の腎機能障害を有する非 HCV 感染被験者に投与した後に評価されました。クレアチニンクリアランス値が 10 ~ 30 mL/min の被験者では、対照被験者 (クレアチニンクリアランスが 90 mL/min を超える) と比較して、平均 AUCtf 値が 3 倍高かった。クレアチニンクリアランス値が 30 ~ 60 mL/min の被験者では、対照被験者と比較して AUCtf が 2 倍でした。 AUCtf の増加は、これらの被験者の腎および非腎クリアランスの減少によるものと思われます。第 3 相有効性試験には、クレアチニン クリアランス値が 50 mL/分を超える被験者が含まれていました。リバビリンの複数回投与の薬物動態は、腎機能障害のある患者では正確に予測できません。リバビリンは血液透析では効果的に除去されません。
クレアチニンクリアランスが 50 mL/min 未満の患者は、REBETOL で治療すべきではありません [ 禁忌 ]。
肝機能障害
肝機能障害の影響は、リバビリン(600 mg)の単回経口投与後に評価されました。平均 AUCtf 値は、対照被験者と比較した場合、軽度、中等度、または重度の肝機能障害 (Child-Pugh 分類 A、B、または C) の被験者で有意差はありませんでした。しかし、平均 Cmax 値は、肝機能障害の重症度とともに増加し、重度の肝機能障害のある被験者では、対照被験者と比較して 2 倍大きかった。
高齢患者
高齢者の薬物動態評価は実施されていません。
性別
18 人の男性被験者と 18 人の女性被験者の単回投与試験では、臨床的に有意な薬物動態の違いは認められませんでした。
小児患者
5 歳から 16 歳までの慢性 C 型肝炎の小児被験者における REBETOL 200mg カプセルおよび INTRON A の反復投与薬物動態特性を表 12 に要約します。小児科。
REBETOL 経口溶液の完全な薬物動態特性は、小児対象では決定されていません。リバビリン Cmin 値は、小児被験者 (3 歳から 16 歳) における 48 週間の治療中のレベトール経口液剤またはレベトール 200mg カプセルの投与後、同様でした。
3 歳から 17 歳までの C 型慢性肝炎の小児被験者を対象とした臨床試験が実施され、PegIntron および REBETOL (カプセルおよび経口溶液) の薬物動態が評価されました。体表面積を調整した PegIntron の 60 mcg/m²/週の投与を受けた小児被験者では、投与間隔中の曝露の対数変換比推定値は、58% [90% CI: 141%, 177%] 1.5 mcg/kg/週の成人で観察されました。この試験における REBETOL の薬物動態 (用量正規化) は、小児被験者および成人における INTRON A と組み合わせた REBETOL 200mg の以前の試験で報告されたものと同様でした。
リバビリンの吸収に対する食物の影響
レベトールカプセルを高脂肪食(841 kcal、脂肪 53.8 g、タンパク質 31.6 g、炭水化物 57.4 g)と一緒に投与した場合、単回投与の薬物動態試験では、AUCtf と Cmax の両方が 70% 増加しました [ 投薬と管理 ]。
微生物学
作用機序
リバビリンが臨床における抗ウイルス効果に寄与するメカニズムは完全には理解されていません。リバビリンは、組織培養において多くの RNA ウイルスに対して直接的な抗ウイルス活性を示します。リバビリンはいくつかのウイルスのゲノムの変異頻度を増加させ、リバビリン三リン酸は生化学反応で HCV ポリメラーゼを阻害します。
細胞培養における抗ウイルス活性
HCVレプリコンにおけるリバビリンの抗ウイルス活性はよく理解されておらず、リバビリンの細胞毒性のために定義されていません。直接的な抗ウイルス活性は、他の RNA ウイルスの組織培養で観察されています。インターフェロンの抗HCV活性は、自己複製HCV-RNS(HCVレプリコン細胞)またはHCV感染を含む細胞で実証されました。
抵抗
HCV 遺伝子型は、ペグ化組換えヒト インターフェロン/リバビリン療法に対する反応に大きなばらつきを示します。可変応答に関連する遺伝子変化は特定されていません。
交差抵抗
ペグ化/非ペグ化インターフェロンとリバビリン間の交差耐性は報告されていません。
動物毒物学および薬理学
マウスおよびラットにおける長期試験 [18 ~ 24 か月;それぞれ 20 から 75 および 10 から 40 mg/kg/日の用量 (60 kg の成人の体表面積調整に基づく推定ヒト等価用量は、それぞれ 1.67 から 6.25 および 1.43 から 5.71 mg/kg/日; およそヒトのリバビリンの 24 時間最大用量の 0.1 から 0.4 倍)] は、リバビリンの慢性曝露とマウスの血管病変(顕微鏡的出血)の発生率の増加との関係を示しています。ラットでは、対照群で網膜変性が発生したが、リバビリン投与ラットでは発生率が増加した。
出生後に子ラットに 10、25、および 50 mg/kg/日の用量でリバビリンを投与した研究では、50 mg/kg で薬物関連の死亡が発生しました (ヒトにおけるヒト血漿濃度よりも低いラット子の血漿濃度で)。生後 7 日目から 63 日目に投与された子ラットは、すべての投与量で全体的な成長のわずかな用量依存的な減少を示し、その後、体重、頭頂部の長さ、そして骨の長さ。これらの影響は可逆性の証拠を示し、骨に対する組織病理学的影響は観察されませんでした。神経行動または生殖発達に関して、リバビリンの影響は観察されませんでした。
臨床研究
臨床試験 1 では、PegIntron 単剤療法が評価されました。見る この試験に関する情報の PegIntron ラベル。
REBETOL/PegIntron 併用療法
成人対象
スタディ 2
無作為化試験では、2 つの PegIntron/REBETOL 200mg レジメン [PegIntron 1.5 mcg/kg を週 1 回皮下投与/REBETOL 800 mg を毎日経口投与 (分割投与)] による治療が比較されました。 PegIntron 1.5 mcg/kg を週 1 回皮下投与して 4 週間、その後 0.5 mcg/kg を週 1 回皮下投与して 44 週間/レベトール 1000 または 1200 mg を 1 日 1 回経口投与 (分割投与)] イントロン A [3 MIU を週 3 回皮下投与/レベトール 1000 またはC型慢性肝炎の成人1530人を対象に、1日1200mgを経口で(分割投与)]インターフェロン未使用の被験者を48週間治療し、治療後24週間追跡しました。適格な被験者は、代償性肝疾患、検出可能な HCV-RNA、ALT の上昇、および慢性肝炎と一致する肝臓の組織病理学を有していました。
治療に対する応答は、治療後 24 週間で検出されない HCV-RNA として定義されました (表 13 を参照)。 PegIntron 1.5 mcg/kg およびリバビリン 800 mg の用量に対する反応率は、INTRON A/REBETOL に対する反応率よりも高かった (表 13 を参照)。PegIntron 1.5 → 0.5 mcg/kg/REBETOL 200 mg に対する反応率は本質的に同じであったINTRON A/REBETOL への応答として (データは示していません)。
ウイルス量に関係なく、ウイルス遺伝子型 1 の被験者は、他のウイルス遺伝子型の被験者と比較して、PegIntron (1.5 mcg/kg)/REBETOL (800 mg) に対する反応率が低かった。予後不良因子(遺伝子型 1 と高ウイルス量)の両方を有する被験者の奏効率は 30%(78/256)であったのに対し、INTRON A/REBETOL 併用療法の奏効率は 29%(71/247)でした。
体重が少ない被験者ほど、有害反応率が高くなる傾向があった[参照 有害反応 であり、体重の大きい被験者よりも高い反応率を示しています。治療群間の反応率の差は、体重による実質的な変化はありませんでした。
PegIntron/REBETOL 併用療法による治療反応率は、男性で 49%、女性で 56% でした。回答率は、アフリカ系アメリカ人とヒスパニック系被験者で低く、白人と比較してアジア人で高かった。アフリカ系アメリカ人は白人に比べて予後不良因子の割合が高かったが、調査された非白人の数 (全体の 11%) は、この試験で予後因子を調整した後の応答率の違いについて意味のある結論を出すには不十分であった.
被験者の 68% で治療の前後に肝生検が得られました。ベースラインと比較して、すべての治療グループの被験者の約 3 分の 2 で、炎症がわずかに減少したことが観察されました。
スタディ 3
米国のコミュニティベースの大規模な試験では、慢性 C 型肝炎の 4913 人の被験者が無作為に割り付けられ、PegIntron 1.5 mcg/kg を週 1 回、800 ~ 1400 mg の REBETOL 200 mg 用量と組み合わせて皮下投与されました (体重ベースの投与 [WBD]) または遺伝子型に基づいて 24 週間または 48 週間、毎日 800 mg (一定量) 経口 (分割用量)。治療に対する反応は、治療後 24 週間で検出されない HCV-RNA (検出下限 125 IU/mL のアッセイに基づく) として定義されました。
PegIntron 1.5 mcg/kg と REBETOL 800 ~ 1400 mg による治療は、1 日 800 mg の均一な REBETOL と組み合わせた PegIntron と比較して、より高い持続的なウイルス学的反応をもたらしました。体重が 105 kg を超える被験者は、WBD で最大の利益を得ましたが、体重が 85 ~ 105 kg を超える被験者でもわずかな利益が観察されました (表 14 を参照)。体重が 85 kg を超える被験者における WBD の利点は、HCV 遺伝子型 1 ~ 3 で観察されました。他の遺伝子型に関して結論を出すには不十分なデータしか入手できませんでした。 WBDの使用により、貧血の発生率が増加しました[参照 有害反応 ]。
研究 3 では、体重が 65 kg を超える合計 1,552 人の被験者が遺伝子型 2 または 3 を有し、24 週間または 48 週間の治療に無作為に割り付けられました。治療期間が長くなっても、追加の利点は観察されませんでした。
スタディ 4
大規模な無作為化試験では、48 週間の治療の安全性と有効性を 2 つの PegIntron/REBETOL 200 mg レジメン [PegIntron 1.5 mcg/kg および 1 mcg/kg を週 1 回皮下投与し、どちらも REBETOL 800 ~ 1400 mg PO を毎日 (2 回に分けて) と組み合わせて比較しました。 C 型慢性肝炎遺伝子型 1 の未治療成人 3,070 人を対象に、ペガシス 180 mcg を週 1 回皮下投与し、コペガス 1,000 ~ 1,200 mg を毎日 (2 回に分けて) PO と併用した。 HCV-RNA またはベースラインからの 2 log10 以上の減少) 12 週目は、治療中止の基準でした。 SVRは、治療後24週間で検出されないHCV-RNA(Roche COBAS TaqManアッセイ、27IU/mLの定量下限)として定義された(表15参照)。
全体的な SVR 率は、3 つの治療群間で類似していました。治療群に関係なく、SVR 率は、予後因子が不良な被験者で低かった。しかし、PegIntron (1.5 mcg/kg)/REBETOL または Pegasys/Copegus に無作為に割り付けられた予後不良因子を持つ被験者は、PegIntron 1 mcg/kg/REBETOL に無作為に割り付けられた同様の被験者と比較して、より高い SVR 率を達成しました。 PegIntron 1.5 mcg/kg と REBETOL の投与量について、次の予後因子を持つ被験者と持たない被験者の SVR 率は次のとおりでした: 肝硬変 (10% 対 42%)、正常な ALT レベル (32% 対 42%)、ベースラインのウイルス600,000 IU/mL を超える負荷 (35% 対 61%)、40 歳以上 (38% 対 50%)、アフリカ系アメリカ人 (23% 対 44%)。 PegIntron (1.5 mcg/kg)/REBETOL 200 mg を投与された治療 12 週目に HCV-RNA が検出されなかった被験者では、SVR 率は 81% (328/407) でした。
研究 5 - 治療失敗例における REBETOL/PegIntron 併用療法
非比較試験では、αインターフェロン/リバビリンの組み合わせによる以前の治療に失敗した中等度から重度の線維症の2293人の被験者が、体重調整されたリバビリンと組み合わせて、1.5 mcg / kgのPegIntronを週1回皮下投与して再治療されました。適格な被験者には、以前の非応答者(最低12週間の治療の終わりにHCV-RNA陽性であった被験者)および以前の再発者(最低12週間の治療の終わりにHCVRNA陰性であり、その後治療後に再発した被験者)が含まれていましたファローアップ)。 12 週目に陰性だった被験者は 48 週間治療を受け、治療後 24 週間追跡されました。治療に対する反応は、治療後 24 週間で HCV-RNA が検出されないことと定義されました (研究ベースのテストを使用して測定、検出限界 125 IU/mL)。全体の奏効率は 22% (497/2293) (99% CI: 19.5, 23.9) でした。次の特徴を持つ被験者は、再治療の恩恵を受ける可能性が低くなりました:以前の非反応、以前のペグ化インターフェロン治療、重大な架橋線維症または肝硬変、および遺伝子型1感染。
ベースライン特性による再治療持続ウイルス学的反応率を表16に要約する。
治療 12 週目に HCV-RNA が検出されないことは、SVR の強力な予測因子でした。この試験では、1470 人 (64%) の被験者が治療 12 週目に検出不能な HCV-RNA を達成せず、治療反応が不十分であったため、長期治療試験への登録が提案されました。治療 12 週目に HCV-RNA が検出されなかった 823 人 (36%) の被験者のうち、遺伝子型 1 に感染した被験者の SVR は 48% (245/507) で、線維化スコア (F4-F2) による反応の範囲は39~55%。ジェノタイプ 2/3 に感染し、治療 12 週目に HCV-RNA が検出されなかった被験者の全体的な SVR は 70% (196/281) で、線維症スコア (F4-F2) による反応の範囲は 60-83% でした。すべての遺伝子型について、線維化スコアが高いほど、SVR を達成する可能性が低くなった。
小児科
型慢性肝炎が代償され、HCV-RNA が検出可能な未治療の 3 歳から 17 歳の小児被験者は、HCV 遺伝子型とベースラインのウイルスに基づいて、1 日あたり 15 mg/kg の REBETOL と 60 mcg/m² の PegIntron で週 1 回、24 週間または 48 週間治療されました。ロード。すべての被験者は、治療後24週間追跡されました。合計 107 人の被験者が治療を受け、そのうち 52% が女性、89% が白人、67% が HCV 遺伝子型 1 に感染していました。 600,000 以上の HCV-RNA を持つ遺伝子型 1、4 または遺伝子型 3 に感染した被験者IU/mL は 48 週間の治療を受けましたが、HCV-RNA が 600,000 IU/mL 未満のジェノタイプ 2 またはジェノタイプ 3 に感染した患者は 24 週間の治療を受けました。試行結果を表 17 にまとめます。
レベトール/イントロンA併用療法
成人対象
以前に未治療の被験者
代償性C型慢性肝炎および検出可能なHCV-RNA(研究に基づくRT-PCRアッセイを使用して中央研究所によって評価された)を有し、以前にアルファインターフェロン療法で治療されていない成人が、2つの多施設二重盲検試験(米国および国際)に登録されました。 REBETOL 200mg カプセルを 1 日 1200 mg (体重が 75 kg 以下の場合は 1 日 1000 mg) および INTRON A 3 MIU を週 3 回、または INTRON A とプラセボを 24 または 48 週間、その後 24 週間投与するように無作為に割り付けられました。オフセラピーのフォローアップ。国際試験には、24 週間の INTRON A とプラセボ治療群は含まれていませんでした。米国の試験には、ベースラインで 67% が男性、89% がコーカサス人で平均 Knodell HAI スコア (I+II+III) が 7.5、72% が遺伝子型 1 である 912 人の被験者が登録されました。ヨーロッパ、イスラエルで実施された国際試験、カナダ、およびオーストラリアでは、799 人の被験者が登録されました (65% が男性、95% が白人、平均クノデル スコア 6.8、および 58% 遺伝子型 1)。
試行結果を表 18 にまとめます。
REBETOL/INTRON A 治療の 24 週目までに研究ベースのアッセイの検出限界を下回る HCV-RNA を達成しなかった被験者のうち、追加の 24 週間の併用治療に反応したのは 5% 未満でした。
レベトール/イントロン A 療法で治療された HCV ジェノタイプ 1 の被験者のうち、HCV-RNA が研究に基づくアッセイの検出限界を 24 週間下回った場合、48 週間の治療に無作為に割り付けられた被験者は、24 週間の被験者と比較してより高いウイルス学的反応を示しました。週間治療グループ。 24 週間と比較して、48 週間の REBETOL/INTRON A 療法に無作為に割り付けられた HCV 非遺伝子型 1 の被験者の反応率の増加は観察されませんでした。
再発被験者
代償性C型慢性肝炎および検出可能なHCV-RNA(研究ベースのRT-PCRアッセイを使用して中央研究所によって評価された)を有し、1または2コースのインターフェロン療法(異常な血清ALTレベルとして定義される)後に再発した被験者は、2つに登録されました多施設、二重盲検試験 (米国および国際) および REBETOL 1200 mg/日 (体重 ≤ 75 kg の被験者には 1000 mg/日) および INTRON A 3 MIU を週 3 回、または INTRON A とプラセボを 24 週間投与するように無作為化された後、 24週間のオフセラピーフォローアップ。米国の試験では、ベースラインで 67% が男性、92% がコーカサス人で平均 Knodell HAI スコア (I+II+III) が 6.8、遺伝子型 1 が 58% の 153 人の被験者が登録されました。ヨーロッパ、イスラエルで実施された国際試験、カナダ、およびオーストラリアでは、192 人の被験者が登録されました (64% が男性、95% が白人、平均クノデル スコア 6.6、および 56% が遺伝子型 1)。試験結果を表 19 にまとめます。
ウイルス学的および組織学的反応は、未治療の試験と再発試験の両方で、男性と女性の被験者間で同様でした。
小児科
C 型代償性慢性肝炎および検出可能な HCV-RNA を有する 3 歳から 16 歳の小児被験者 (研究に基づく RT-PCR アッセイを使用して中央検査室で評価) は、1 日あたり 15 mg/kg の REBETOL および INTRON A 3 MIU/で治療されました。 m² を週 3 回 48 週間続け、続いて 24 週間のオフセラピー フォローアップを行います。合計 118 人の被験者が治療を受け、そのうち 57% が男性、80% が白人、78% が遺伝子型 1 でした。カプセル。
試験結果を表 20 にまとめます。
ウイルス量に関係なく、ウイルス遺伝子型 1 の被験者は、遺伝子型非 1 の被験者と比較して、INTRON A/REBETOL 併用療法に対する反応率が 36% 対 81% と低かった。予後不良因子 (遺伝子型 1 とウイルス量の高値) の両方を持つ被験者の奏効率は 26% (13/50) でした。
患者情報
情報が提供されていません。を参照してください。 警告と注意事項 セクション。