ヘアトリートメント: Avodart 0.5mg Dutasteride 使用法、副作用および投与量。 オンライン薬局の価格。 処方箋不要のジェネリック医薬品。
アボダート 0.5mg とは何ですか?
アボダート 0.5mg は、前立腺肥大症 (良性前立腺肥大症) の症状を治療するために使用される処方薬です。アボダートは、単独で使用することも、他の薬と併用することもできます。
アボダート 0.5mg は、5-アルファ還元酵素阻害剤と呼ばれる種類の薬物に属しています。
アボダート 0.5mg が子供に安全で効果的かどうかはわかっていません。
アボダートの副作用は何ですか?
アボダート 0.5mg の副作用には次のようなものがあります。
- 蕁麻疹、
- 呼吸困難、
- 顔やのどの腫れ、
- 熱、
- 喉の痛み、
- 燃える目、
- 皮膚の痛みと
- 水ぶくれとはがれを伴う赤または紫の皮膚発疹
上記の症状がある場合は、すぐに医療機関を受診してください。
アボダート 0.5mg の最も一般的な副作用は次のとおりです。
- 性欲減退(性欲)、
- 性交時に放出される精液の量が減少し、
- インポテンス(勃起が得られない、または勃起を維持できない)、および
- 乳房の圧痛または拡大
気になる副作用や治らない副作用がある場合は、医師に相談してください。
これらは、Avodart の考えられるすべての副作用ではありません。詳細については、医師または薬剤師にお尋ねください。
副作用に関する医学的アドバイスについては、医師に連絡してください。 1-800-FDA-1088 で副作用を FDA に報告できます。
説明
アボダート 0.5mg は、テストステロンを DHT に変換する細胞内酵素であるステロイド 5 α-レダクターゼのタイプ 1 およびタイプ 2 アイソフォームの両方の選択的阻害剤である合成 4-アザステロイド化合物です。
デュタステリドは、(5α,17β)-N-{2,5 ビス(トリフルオロメチル)フェニル}-3-オキソ-4-アザアンドロスト-1-エン-17-カルボキサミドと化学的に指定されています。デュタステリドの実験式は C27H30F6N2O2 で、分子量は 528.5 で、次の構造式を持っています。
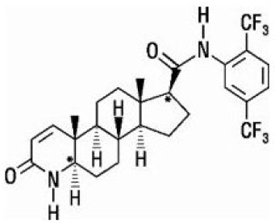
デュタステリドは白色から淡黄色の粉末で、融点は 242°~250°C です。エタノール (44 mg/mL)、メタノール (64 mg/mL)、ポリエチレングリコール 400 (3 mg/mL) に溶けますが、水には溶けません。
経口投与される各アボダート 0.5mg ソフト ゼラチン カプセルには、カプリル酸/カプリン酸のモノジグリセリドとブチル化ヒドロキシトルエンの混合物に溶解した 0.5 mg のデュタステリドが含まれています。カプセル シェル内の不活性賦形剤は、酸化鉄 (黄色)、ゼラチン (認定された BSE フリーの牛由来)、グリセリン、および二酸化チタンです。ソフトゼラチンカプセルは、食用の赤インクで印刷されています。
適応症
単剤療法
アボダート (デュタステリド) ソフト ゼラチン カプセルは、前立腺肥大症の男性における症候性良性前立腺肥大症 (BPH) の治療に適応されます。
- 症状を改善し、
- 急性尿閉(AUR)のリスクを軽減し、
- BPH 関連の手術が必要になるリスクを軽減します。
αアドレナリン拮抗薬との併用
アボダートとα-アドレナリンアンタゴニストであるタムスロシンとの併用は、前立腺肥大症の男性における症候性BPHの治療に適応されます。
使用制限
アボダート 0.5mg は、前立腺がんの予防には承認されていません。
投薬と管理
カプセルの内容物と接触すると、口腔咽頭粘膜が刺激される可能性があるため、カプセルは丸ごと飲み込み、噛んだり開けたりしないでください。アボダートは食事の有無にかかわらず投与できます。
単剤療法
アボダートの推奨用量は、1 日 1 回 1 カプセル (0.5 mg) です。
αアドレナリン拮抗薬との併用
アボダート 0.5mg の推奨用量は、1 カプセル (0.5 mg) を 1 日 1 回、タムスロシン 0.4 mg を 1 日 1 回服用することです。
供給方法
剤形と強度
0.5 mg、不透明、くすんだ黄色のゼラチン カプセルで、片面に赤インクで「GX CE2」と刻印されています。
保管と取り扱い
アヴォダート ソフト ゼラチン カプセル 0.5 mg は、楕円形で不透明なくすんだ黄色のゼラチン カプセルで、「GX CE2」と刻印され、片面に赤の食用インクがあり、30 個入りのボトルに包装されています ( NDC 0173-0712-15)、90( NDC 0173-0712-04) チャイルド レジスタント クロージャー付き。
25°C (77°F) で保管してください。 15° から 30°C (59° から 86°F) まで許容されるエクスカーション [参照 USP制御の室温 ]。
デュタステリドは皮膚から吸収されます。アボダート 0.5 mg カプセルは、デュタステリドの吸収の可能性と、その後の男性胎児の発育に対する潜在的なリスクのため、妊娠中または妊娠の可能性のある女性が取り扱うべきではありません [参照 警告と注意事項 ]。
製造対象: グラクソスミスクライン リサーチ トライアングル パーク。改訂: 2020 年 1 月
副作用
臨床試験の経験
臨床試験はさまざまな条件下で実施されるため、ある医薬品の臨床試験で観察された副作用の発生率を別の医薬品の臨床試験で観察された発生率と直接比較することはできず、実際に観察された発生率を反映していない可能性があります。
AVODART 0.5mgを単剤療法として、またはタムスロシンと併用した臨床試験から:
- アボダート 0.5mg を投与された被験者で報告された最も一般的な副作用は、インポテンス、性欲減退、乳房障害 (乳房の肥大と圧痛を含む)、および射精障害でした。併用療法 (AVODART 0.5mg とタムスロシン) を受けた被験者で報告された最も一般的な副作用は、インポテンツ、性欲減退、乳房障害 (乳房の肥大と圧痛を含む)、射精障害、およびめまいでした。射精障害は、単独療法として AVODART (2%) またはタムスロシン (4%) を受けた被験者と比較して、併用療法 (11%) を受けた被験者で有意に多く発生しました。
- 副作用による試験中止は、AVODART を使用したプラセボ対照試験で、AVODART 0.5mg を投与された被験者の 4%、プラセボを投与された被験者の 3% で発生しました。治験中止に至った最も一般的な副作用はインポテンツ(1%)でした。
- 併用療法を評価する臨床試験では、有害反応による試験中止は、併用療法(AVODART とタムスロシン)を受けた被験者の 6%、および単剤療法として AVODART 0.5mg またはタムスロシンを受けた被験者の 4% で発生しました。試験の中止につながったすべての治療群で最も一般的な副作用は、勃起不全 (1% ~ 1.5%) でした。
単剤療法
前立腺肥大症の 4,300 人以上の男性被験者が無作為に割り当てられ、プラセボまたは毎日 0.5 mg のアボダートを 3 つの同一の 2 年間、プラセボ対照、二重盲検、第 3 相治療試験で受け、その後それぞれ 2 年間の非盲検試験が行われました。拡大。二重盲検治療期間中、2,167 人の男性被験者が AVODART 0.5mg に暴露され、そのうち 1,772 人が 1 年間暴露され、1,510 人が 2 年間暴露されました。非盲検延長を含めると、1,009 人の男性被験者が AVODART 0.5mg に 3 年間暴露され、812 人が 4 年間暴露されました。人口は 47 ~ 94 歳 (平均年齢: 66 歳) で、90% 以上が白人でした。表 1 は、AVODART を投与された被験者の少なくとも 1% で報告され、プラセボを投与された被験者よりも高い発生率で報告された臨床的副作用をまとめたものです。
長期治療(最長4年)
高悪性度前立腺がん
REDUCE 試験は無作為化二重盲検プラセボ対照試験で、血清 PSA が 2.5 ng/mL から 10 ng/mL で、過去 6 か月以内に前立腺生検が陰性であった 50 歳から 75 歳の男性 8,231 人を登録しました。被験者は、無作為にプラセボ (n = 4,126) または 0.5 mg のアボダート (n = 4,105) を最長 4 年間投与されました。平均年齢は 63 歳で、91% が白人でした。被験者は、治療の 2 年目と 4 年目にプロトコールで義務付けられた予定された前立腺生検を受けるか、臨床的に必要な場合は予定外の時間に「理由のある生検」を受けました。アボダートを投与された男性 (1.0%) では、プラセボを投与された男性 (0.5%) と比較して、グリーソンスコア 8-10 の前立腺がんの発生率が高かった [参照 適応症と使用法 、 警告と注意事項 ]。別の5α還元酵素阻害剤(フィナステリド5mg、PROSCAR)を用いた7年間のプラセボ対照臨床試験では、グリーソンスコア8~10の前立腺がんについて同様の結果が観察されました(フィナステリド1.8%対プラセボ1.1%)。
AVODART で治療された前立腺癌患者では、臨床的利益は証明されていません。
生殖器と乳房の病気
AVODART 0.5mgを使用した3つの重要なプラセボ対照BPH試験では、それぞれ4年間の期間で、治療期間の延長に伴う性的有害反応の増加(インポテンス、性欲減退、および射精障害)または乳房障害の証拠はありませんでした.これら 3 つの試験のうち、デュタステリド群で 1 例、プラセボ群で 1 例の乳がんが発生しました。 4 年間の CombAT 試験または 4 年間の REDUCE 試験では、いずれの治療群でも乳がんの症例は報告されませんでした。
デュタステリドの長期使用と男性の乳房腫瘍との関係は現在不明です。
α遮断薬療法(CombAT)との併用
4,800 人を超える BPH の男性被験者が、0.5 mg のアボダート 0.5 mg、0.4 mg のタムスロシン、または併用療法 (0.5 mg アボダートと 0.4 mg タムスロシン) を 4 年間の二重盲検試験で 1 日 1 回投与されるように無作為に割り当てられました。全体で、1,623 人の被験者が AVODART による単剤療法を受けました。 1,611人の被験者がタムスロシンによる単剤療法を受けました。 1,610 人の被験者が併用療法を受けました。人口は 49 ~ 88 歳 (平均年齢: 66 歳) で、88% が白人でした。表 2 は、併用群の被験者の少なくとも 1% で報告され、AVODART 0.5mg またはタムスロシンによる単剤療法を受けた被験者よりも高い発生率で報告された副作用をまとめたものです。
心不全
CombAT では、4 年間の治療後、併用療法グループ (12/1,610; 0.7%) の複合用語心不全の発生率は、いずれかの単独療法グループよりも高かった: AVODART 0.5mg、2/1,623 (0.1%) およびタムスロシン、9/1,611 (0.6%)。複合心不全は、前立腺癌の発症リスクのある男性を対象に AVODART を評価する別の 4 年間のプラセボ対照試験でも調べられました。アボダート 0.5mg を服用した被験者の心不全の発生率は、プラセボを服用した被験者の 0.4% (15/4,126) と比較して、0.6% (26/4,105) でした。両方の試験で心不全の被験者の大部分は、心不全のリスクの増加に関連する併存症を持っていました。したがって、心不全における数値の不均衡の臨床的意義は不明です。アボダート単独またはタムスロシンとの併用と心不全との因果関係は確立されていません。いずれの試験においても、全体的な心血管有害事象の発生率に不均衡は観察されませんでした。
市販後の経験
アボダートの承認後の使用中に、次の副作用が確認されました。これらの反応は不確かな規模の集団から自発的に報告されるため、その頻度を確実に推定したり、薬物曝露との因果関係を確立したりすることは常に可能ではありません.これらの反応は、その深刻さ、報告の頻度、または AVODART との潜在的な因果関係の組み合わせにより、含めるために選択されました。
免疫系疾患
発疹、かゆみ、蕁麻疹、局所浮腫、深刻な皮膚反応、血管性浮腫などの過敏反応。
新生物
男性の乳がん。
精神障害
憂鬱な気分。
生殖器系と乳房の病気
睾丸の痛みと睾丸の腫れ。
薬物相互作用
シトクロム P450 3A 阻害剤
デュタステリドは、シトクロム P450 (CYP)3A4 および CYP3A5 アイソザイムによってヒトで広範囲に代謝されます。デュタステリドに対する強力な CYP3A4 阻害剤の効果は研究されていません。薬物間相互作用の可能性があるため、強力な慢性 CYP3A4 酵素阻害剤 (リトナビルなど) を服用している患者にアボダートを処方する場合は注意が必要です [ 臨床薬理学 ]。
α-アドレナリン拮抗薬
タムスロシンまたはテラゾシンと組み合わせたアボダートの投与は、どちらのアルファアドレナリン拮抗薬の定常状態の薬物動態にも影響を与えません。デュタステリドの薬物動態パラメーターに対するタムスロシンまたはテラゾシンの投与の影響は評価されていません。
カルシウムチャネル拮抗薬
ベラパミルまたはジルチアゼムの同時投与は、デュタステリドのクリアランスを減少させ、デュタステリドへの暴露を増加させます。デュタステリド曝露の変化は、臨床的に重要であるとは考えられていません。用量調整は推奨されない[参照 臨床薬理学 ]。
コレスチラミン
アボダート 0.5mg を 5mg 単回投与し、その 1 時間後に 12g のコレスチラミンを投与しても、デュタステリドの相対的バイオアベイラビリティーに影響はありません。 臨床薬理学 ]。
ジゴキシン
アボダート 0.5mg は、1 日 0.5mg の用量で 3 週間併用投与した場合、ジゴキシンの定常状態の薬物動態を変化させません。 臨床薬理学 ]。
ワルファリン
アボダート 0.5 mg/日をワルファリンと 3 週間併用投与しても、S-または R-ワルファリン異性体の定常状態の薬物動態は変化せず、プロトロンビン時間に対するワルファリンの効果も変化しません。 臨床薬理学 ]。
警告
の一部として含まれています 予防 セクション。
予防
前立腺特異抗原 (PSA) に対する影響と前立腺癌検出における PSA の使用
臨床試験では、アボダート 0.5 mg は、治療後 3 ~ 6 か月以内に血清 PSA 濃度を約 50% 低下させました。この減少は、症候性 BPH の被験者の PSA 値の全範囲にわたって予測可能でしたが、個人によって異なる場合があります。アボダート 0.5mg はまた、前立腺癌の存在下で血清 PSA の低下を引き起こす可能性があります。アボダート 0.5mg を服用している男性の一連の PSA を解釈するには、治療開始から少なくとも 3 か月後に新しい PSA ベースラインを確立し、その後定期的に PSA を監視する必要があります。 AVODART の服用中に最低 PSA 値からの増加が確認された場合は、前立腺がんの存在を示している可能性があり、5 α還元酵素阻害剤を服用していない男性の PSA 値がまだ正常範囲内にある場合でも、評価する必要があります。 AVODART に準拠していないと、PSA テストの結果にも影響を与える可能性があります。
アボダート 0.5mg で 3 か月以上治療を受けた男性の単独の PSA 値を解釈するには、未治療の男性の正常値と比較して PSA 値を 2 倍にする必要があります。 AVODART の影響下でも、遊離総 PSA 比 (遊離 PSA パーセント) は一定のままです。臨床医が AVODART を受けている男性の前立腺癌の検出の補助としてパーセント遊離 PSA を使用することを選択した場合、その値を調整する必要はないようです。
デュタステリドとタムスロシンの同時投与は、デュタステリド単独療法と同様の血清 PSA の変化をもたらしました。
高悪性度前立腺がんのリスク増加
前立腺がんの生検が以前に陰性であり、ベースライン PSA が 2.5 ng/mL から 10.0 ng/mL の間である 50 歳から 75 歳の男性で、4 年間のデュタステリドによる前立腺がんイベントの減少 (REDUCE) 試験でアボダートを服用した場合、プラセボを服用している男性と比較して、グリーソン スコア 810 の前立腺がんの発生率が高い (AVODART 1.0% 対 プラセボ 0.5%) [参照 適応症と使用法 、 有害反応 ]。別の5α還元酵素阻害剤(フィナステリド5mg、PROSCAR)を用いた7年間のプラセボ対照臨床試験では、グリーソンスコア8~10の前立腺がんについて同様の結果が観察されました(フィナステリド1.8%対プラセボ1.1%)。
α-レダクターゼ阻害剤は、高悪性度前立腺がんの発症リスクを高める可能性があります。前立腺容積を減少させる5α-レダクターゼ阻害剤の効果または試験関連の要因がこれらの試験の結果に影響を与えたかどうかは確立されていません.
他の泌尿器疾患の評価
アボダート 0.5mg による治療を開始する前に、同様の症状を引き起こす可能性のある他の泌尿器疾患について考慮する必要があります。さらに、前立腺肥大症と前立腺がんが共存している可能性があります。
妊娠中の女性におけるアボダート 0.5 mg の経皮暴露 - 男性胎児へのリスク
妊娠中または妊娠している可能性のある女性は、アボダート カプセルを扱わないでください。デュタステリドは皮膚から吸収される可能性があり、意図しない胎児への曝露や男性胎児への潜在的なリスクをもたらす可能性があります.妊婦が漏れたデュタステリド カプセルに接触した場合は、接触部分を直ちに石鹸と水で洗浄する必要があります。 特定の集団での使用 ]。デュタステリドは、動物実験に基づいて皮膚から吸収されます [参照 非臨床毒性学 ]。
献血
AVODART の投与を受けている男性は、最後の投与から少なくとも 6 か月が経過するまでは献血しないでください。この延期期間の目的は、デュタステリドが妊娠中の女性の輸血レシピエントに投与されるのを防ぐことです。
精液特性への影響
デュタステリド 0.5 mg/日が精液特性に及ぼす影響は、52 週間の治療期間と 24 週間の治療後のフォローアップ期間を通じて、健康な男性で評価されました。 52 週の時点で、プラセボと比較して、デュタステリド治療により、総精子数、精液量、および精子の運動性が平均的に減少しました。総精子数への影響は、24 週間の追跡調査後も元に戻すことはできませんでした。精子濃度と精子の形態は影響を受けず、すべての精液パラメーターの平均値はすべての時点で正常範囲内にとどまりました。個々の患者の生殖能力に対するデュタステリドの精液特性への影響の臨床的意義は知られていない[ 特定の集団での使用 ]。
患者相談情報
患者に、FDA 承認の患者ラベル ( 患者情報 )。
PSAモニタリング
個人差はありますが、AVODART は治療後 3 ~ 6 か月以内に血清 PSA 値を約 50% 低下させることを患者に伝えてください。 PSA スクリーニングを受けている患者の場合、AVODART による治療中の PSA レベルの上昇は、前立腺がんの存在を示す可能性があるため、医療提供者によって評価されるべきです [ 警告と注意事項 ]。
高悪性度前立腺がんのリスク増加
アボダート 0.5 mg を含む 5 種類の α-レダクターゼ阻害剤 (BPH 治療に適応) で治療された男性では、これらの使用を検討した試験でプラセボで治療された男性と比較して、高悪性度前立腺がんが増加したことを患者に知らせます。前立腺がんのリスクを軽減するための薬 [参照 適応症と使用法 、 警告と注意事項 、 有害反応 ]。
妊娠中または妊娠の可能性のある女性におけるアボダートの経皮暴露 - 男性胎児へのリスク
デュタステリドが吸収される可能性があり、その後、男性胎児の発育にリスクが生じる可能性があるため、妊娠中または妊娠の可能性のある女性は、AVODART 0.5mg カプセルを取り扱ってはならないことを患者に伝えてください。デュタステリドは皮膚から吸収される可能性があり、意図しない胎児への曝露を引き起こす可能性があります.妊娠中または妊娠している可能性のある女性が、漏れたアボダート 0.5mg カプセルに接触した場合は、接触部分を石鹸と水で直ちに洗浄する必要があります [参照 警告と注意事項 、 特定の集団での使用 ]。
精液パラメーターへの影響
男性には、アボダート 0.5mg が精子の特性に影響を与える可能性があるが、生殖能力への影響は不明であることを助言する [ 警告と注意事項 、 特定の集団での使用 ]。
献血
アボダート 0.5mg を投与された男性には、妊娠中の女性が輸血によってデュタステリドを投与されるのを防ぐために、最終投与から少なくとも 6 か月後までは献血しないことを伝えてください。 警告と注意事項 ]。デュタステリドの血清レベルは、治療終了後 4 ~ 6 か月間検出可能です [参照 臨床薬理学 ]。
AVODART は、GSK グループ企業が所有またはライセンス供与している商標です。
リストされているその他のブランドは、それぞれの所有者が所有またはライセンス供与している商標であり、GSK グループ企業が所有またはライセンス供与しているものではありません。これらのブランドのメーカーは、GSK グループの企業またはその製品と提携しておらず、推奨もしていません。
非臨床毒性学
発がん、突然変異誘発、生殖能力の障害
発がん
2 年間の発がん性試験が B6C3F1 マウスで、雄では 3、35、250、および 500 mg/kg/日、雌では 3、35、および 250 mg/kg/日の用量で実施された。良性肝細胞腺腫の発生率の増加は、メスのマウスのみで 250 mg/kg/日 (0.5 mg の 1 日用量の MRHD の 290 倍) で認められました。 3 つの主要なヒト代謝物のうち 2 つがマウスで検出されています。マウスにおけるこれらの代謝産物への曝露は、ヒトよりも低いか、または不明です。
Han Wistar ラットの 2 年間の発がん性試験では、雄で 1.5、7.5、および 53 mg/kg/日、雌で 0.8、6.3、および 15 mg/kg/日の用量で、ライディッヒ細胞が増加しました。 MRHDの135倍(53mg/kg/日以上)で精巣の腺腫。ライディッヒ細胞過形成の発生率の増加は、MRHD の 52 倍で存在しました (7.5 mg/kg/日以上の雄ラット用量)。ライディッヒ細胞の増殖変化と循環黄体形成ホルモンレベルの増加との間の正の相関は、5α-レダクターゼ阻害剤で実証されており、5α-レダクターゼ阻害後の視床下部-下垂体-精巣軸への影響と一致しています。腫瘍形成用量では、ラットの黄体形成ホルモンレベルが 167% 増加しました。この研究では、予想される臨床暴露の約 1 ~ 3 倍で、主要なヒト代謝産物の発がん性がテストされました。
突然変異誘発
デュタステリドの遺伝毒性は、細菌変異誘発試験(エイムズ試験)、チャイニーズハムスター卵巣細胞における染色体異常試験、およびラットにおける小核試験で試験されました。結果は、親薬の遺伝毒性の可能性を示しませんでした。 2 つの主要なヒト代謝物も、エイムズ試験または簡略化されたエイムズ試験のいずれかで陰性でした。
生殖能力の障害
平均血清濃度に基づいて MRHD の 0.1 倍のデュタステリド (0.05 mg/kg/日以上の動物用量を最大 31 週間) で性的に成熟した雄ラットを処理すると、すべての用量で受胎能が用量依存的および時間依存的に低下した;精巣上体尾部 (絶対) 精子数は減少したが、精子濃度は減少しなかった (50 および 500 mg/kg/日)。精巣上体、前立腺、および精嚢の重量の減少;および、父性毒性の非存在下でのすべての用量での生殖器における微視的変化(精巣上体の尿細管上皮の細胞質空胞化および/または上皮の細胞質含有量の減少、前立腺および精嚢の分泌活性の低下と一致する)。妊孕性への影響は、すべての用量で回復週 6 までに逆転し、精子数は 14 週間の回復期間の終わりには正常でした。微視的な変化は、MRHD の 0.1 倍の回復 14 週目ではもはや存在せず、残りの治療群では部分的に回復しました。低レベルのデュタステリド (0.6 から 17 ng/mL) が、治療を受けた雄ラットと交配させた未治療の雌ラット (10 から 500 mg/kg/日で 29 から 30 週間) の血清で検出され、これは MRHD の 16 から 110 倍である。平均血清濃度。メスのラットでデュタステリドの検出可能な血中レベルが観察されたにもかかわらず、未処理のメスのラットを処理したオスのラットと交配させたオスの子孫では、メス化は発生しませんでした。
妊娠初期から交尾の 4 週間前に投与したメスのラットの受胎能試験では、デュタステリドを 0.05、2.5、12.5、および 30 mg/kg/日の用量で経口投与すると、再吸収の増加と女性化により同腹児のサイズが減少しました。母体毒性 (体重増加の減少) の存在下で、平均血清濃度に基づいて MRHD (2.5 mg/kg/日以上の動物用量) の 2 ~ 10 倍の雄胎児 (肛門性器間距離の減少)。胎児の体重も、母体毒性がない場合、平均血清濃度に基づいて MRHD の約 0.02 倍 (0.05 mg/kg/日以上のラット用量) で減少し、無影響レベルではなかった。
特定の集団での使用
妊娠
リスクの概要
アボダート 0.5mg は、男性の胎児に害を及ぼす可能性があるため、妊娠中の使用は禁忌です。 禁忌 ]。 AVODART は、女性への使用は適応されていません。
アボダート 0.5mg は、男性生殖器の正常な発達に必要なホルモンであるジヒドロテストステロン (DHT) へのテストステロンの変換を妨げる 5 アルファ還元酵素阻害剤です。雄胎児の生殖器の異常は、この変換の阻害の予想される生理学的結果です。これらの結果は、遺伝的 5 α-レダクターゼ欠損症の男児で観察された結果と類似しています。
米国の一般集団では、臨床的に認識された妊娠における主要な先天性欠損症および流産の推定背景リスクは、それぞれ 2% ~ 4% および 15% ~ 20% です。
動物の繁殖研究では、器官形成期のラットまたはウサギにデュタステリドを 1 日 0.5 mg の最大推奨ヒト用量 (MRHD) 未満で与えた場合、母体毒性がない場合、オスの子孫の外性器の正常な発達を阻害しました。 MRHD の 15 倍で、妊娠期間の延長、生殖器官の重量の減少、雄の子孫の思春期の遅延がラットで観察され、影響のないレベルは MRHD の 0.5 mg 未満でした。ウサギの胎盤重量の増加も観察され、無影響レベルは毎日 0.5 mg の MRHD 未満でした ( データ )。
デュタステリドは人間の精液に分泌されますが、人間の女性パートナーの薬物濃度は、動物実験で男性生殖器の異常を引き起こす濃度の約 100 分の 1 です ( データ )。器官形成期にヒトのメスのパートナーが曝露すると推定されるレベルと同等またはそれ以上の血中濃度をサルに投与した場合、オスの子孫の外性器は悪影響を受けませんでした。メスのラットで検出可能な血中レベルのデュタステリドが観察されたにもかかわらず、未処理のメスのラットを処理したオスのラットと交配させたオスの子孫では、メス化は発生しませんでした。 非臨床毒性学 ]。
データ
ヒューマンデータ
治療を受けた男性で測定されたデュタステリドの最高精液濃度は 14 ng/mL でした。デュタステリドは精液中に検出されますが、体重 50 kg の女性が 5 mL の精液にさらされ、100% 吸収されると仮定すると、女性の精液中のデュタステリド血中濃度は約 0.0175 ng/mL と予想されます。この濃度は、動物実験で男性生殖器の異常を引き起こす血中濃度の約 100 分の 1 です。デュタステリドは、人の精液に高度に結合したタンパク質 (96% 以上) であるため、膣吸収に利用できるデュタステリドの量が減少する可能性があります。
動物データ
ラットの胚・胎児発生研究では、デュタステリドを MRHD の 0.5 mg/日 (男性の平均血中濃度に基づく) の 10 分の 1 で経口投与すると、胎児の男性生殖器が女性化されました (0.05 mg で肛門性器の距離が減少しました)。 /kg/日、母体毒性がない場合、無影響レベルの欠如)。さらに、2.5 mg/kg/日以上 (MRHD の約 15 倍) の用量で処理された母動物の胎児では、乳頭の発達、尿道下裂、および包皮腺の拡張が発生しました。母体毒性(体重増加の減少)の存在下での胎児体重の減少とそれに伴う骨化の遅延が、MRHD の約 15 倍の母体暴露(2.5 mg/kg/日以上の用量)で観察された。 30 mg/kg/日 (MRHD の約 111 倍) を投与した母動物では、死産児の増加が観察され、無影響レベルは 12.5 mg/kg/日でした。
ウサギの胚・胎児発生研究では、男性の平均血中濃度に基づいて MRHD の 28 倍の用量 (30 mg/kg/日以上の用量) が、妊娠 7 日目から 29 日目 (器官形成期および後期) に経口投与されました。外性器の発達期)。胎児の生殖乳頭の組織学的評価は、雄胎児の女性化の証拠、ならびに母体毒性の非存在下でのすべての用量での頭蓋骨の融合および胎盤重量の増加を明らかにした。妊娠中 (器官形成および外性器発生後期 [妊娠 6 ~ 29 日]) に MRHD の 0.3 倍 (0.05 mg/kg/日以上の用量、また、雄胎児の生殖器の女性化の証拠と、母体毒性がない場合のすべての用量での胎盤重量の増加も示した。
胚・胎児発生研究では、妊娠中のアカゲザルは、器官形成中 (妊娠 20 日から 100 日) に、人間の女性パートナーの推定デュタステリド曝露と同等またはそれ以上のデュタステリド血中濃度に静脈内曝露されました。デュタステリドは、妊娠 20 ~ 100 日目 (器官形成期) に 400、780、1,325、または 2,010 ng/日 (12 匹のサル/グループ) の用量で投与されました。サルの子孫のオスの外性器の女性化は観察されなかった。胎児副腎重量の減少、胎児前立腺重量の減少、および胎児卵巣および精巣重量の増加が、試験した最高用量で観察された。治療を受けた男性で測定されたデュタステリドの最高精液濃度 (14 ng/mL) に基づくと、サルでのこれらの用量は、50 kg のヒト女性がデュタステリドから毎日 5 mL の精液にさらされる可能性のある最大暴露量の最大 16 倍に相当します。 100% の吸収を仮定して、処理された男性。この研究でサルに投与された用量レベル (ng/kg ベース) は、女性が精液を介して潜在的に暴露される公称 (ng/kg) 用量の 32 から 186 倍です。ウサギやアカゲザルが主要なヒト代謝産物を産生するかどうかはわかっていません。
ラットの出生前および出生後の経口発育試験では、雄性器の女性化が観察されました。 AUC の推定値としての男性の平均血中濃度に基づいて、肛門性器間距離の減少が MRHD の 0.05 倍以上 (0.05 mg/kg/日以上) で観察され、無影響レベルはありませんでした。 2.5mg/kg/日以上(MRHDの14倍以上、0.05mg/kg/日で無影響量)で尿道下裂と乳頭発育が認められました。 2.5 mg/kg/日以上の用量でも、親の雌の妊娠期間の延長、雄の子孫の包皮包皮分離までの時間の増加、雌の子孫の膣の開通までの時間の減少、および前立腺と前立腺の減少をもたらしました。雄の子孫における精嚢重量。死産の増加と子孫の新生児生存率の低下が 30 mg/kg/日で認められた (母体毒性 [体重の減少] が存在する場合の MRHD の 102 倍)。
授乳
リスクの概要
AVODART は、女性への使用は適応されていません。母乳中のデュタステリドの存在、母乳で育てられた子供への影響、または乳生産への影響に関する入手可能な情報はありません.
生殖能力のある雌と雄
不妊
男性
デュタステリド 0.5 mg/日の効果は、18 歳から 52 歳までの正常なボランティア (n = 27 デュタステリド、n = 23 プラセボ) を対象に、52 週間の治療と 24 週間の治療後のフォローアップを通じて評価されました。プラセボ群のベースラインからの変化を調整した場合、デュタステリド群では、52週で、総精子数、精液量、および精子運動性のベースラインからの平均減少率は、それぞれ23%、26%、および18%でした。精子濃度と精子の形態は影響を受けませんでした。 24 週間のフォローアップ後、デュタステリド群の総精子数の平均変化率は、ベースラインより 23% 低いままでした。すべての時点でのすべての精液パラメーターの平均値は正常範囲内にとどまり、臨床的に有意な変化の事前定義された基準 (30%) を満たしていませんでしたが、デュタステリド群の 2 人の被験者は、ベースラインから 90% を超える精子数の減少を示しました。 52 週間、24 週間のフォローアップで部分的に回復。個々の患者の生殖能力に対するデュタステリドの精液特性への影響の臨床的意義は知られていない[ 警告と注意事項 ]。
小児用
アボダート 0.5mg は、小児患者への使用は適応されていません。小児患者における安全性と有効性は確立されていません。
高齢者の使用
3 つの臨床試験でアボダート 0.5mg を投与された 2,167 人の男性被験者のうち、60% が 65 歳以上、15% が 75 歳以上でした。これらの被験者と若い被験者の間で、安全性または有効性の全体的な違いは観察されませんでした。他の報告された臨床経験では、高齢患者と若年患者の間の反応の違いは確認されていませんが、一部の高齢者の感受性が高いことは除外できません[参照] 臨床薬理学 ]。
腎障害
腎機能障害のある患者では、アボダート 0.5mg の用量調整は必要ありません。 臨床薬理学 ]。
肝障害
デュタステリドの薬物動態に対する肝障害の影響は研究されていません。デュタステリドは広範囲に代謝されるため、肝障害のある患者では曝露が高くなる可能性があります。しかし、60 人の被験者が毎日 5 mg (治療用量の 10 倍) を 24 週間摂取した臨床試験では、0.5 mg の治療用量で観察されたものと比較して、追加の有害事象は観察されませんでした [ 臨床薬理学 ]。
過剰摂取
ボランティア試験では、最大 40 mg (治療用量の 80 倍) までのデュタステリドを 7 日間単回投与したところ、重大な安全上の懸念はありませんでした。臨床試験では、1 日 5 mg (治療用量の 10 倍) の用量を 60 人の被験者に 6 か月間投与しましたが、0.5 mg の治療用量で見られたものに追加の悪影響はありませんでした。
デュタステリドに特効薬はありません。したがって、過剰摂取が疑われる場合は、デュタステリドの半減期が長いことを考慮して、対症療法および支持療法を適切に行う必要があります。
禁忌
アボダート 0.5mg は、以下での使用が禁忌です。

- 妊娠。デュタステリドの使用は、妊娠中の女性には禁忌です。動物の生殖および発生毒性研究では、デュタステリドは雄胎児の外性器の発生を阻害しました。したがって、妊娠中の女性にアボダート 0.5mg を投与すると、胎児に害を及ぼす可能性があります [参照 警告と注意事項 、 特定の集団での使用 ]。
- AVODARTまたは他の5α-レダクターゼ阻害剤に対する臨床的に重大な過敏症(例、重篤な皮膚反応、血管性浮腫)が以前に示された患者[参照 有害反応 ]。
臨床薬理学
作用機序
デュタステリドは、テストステロンからDHTへの変換を阻害します。 DHT は、前立腺の初期発生とその後の肥大に主に関与するアンドロゲンです。テストステロンは、1 型と 2 型の 2 つのアイソフォームとして存在する酵素 5 α-レダクターゼによって DHT に変換されます。皮膚と肝臓。
デュタステリドは、1 型および 2 型両方の 5 α-レダクターゼ アイソザイムの競合的かつ特異的な阻害剤であり、安定した酵素複合体を形成します。この複合体からの解離は、in vitro および in vivo 条件下で評価されており、非常に遅いです。デュタステリドはヒトアンドロゲン受容体に結合しません。
薬力学
5 α-ジヒドロテストステロンとテストステロンへの影響
DHT の減少に対するデュタステリドの 1 日用量の最大効果は用量依存的であり、1 ~ 2 週間以内に観察されます。デュタステリド 0.5 mg を毎日 1 週間および 2 週間服用した後、血清 DHT 濃度の中央値はそれぞれ 85% および 90% 減少しました。デュタステリド 0.5 mg/日で 4 年間治療された BPH 患者では、血清 DHT の減少の中央値は 1 年で 94%、2 年で 93%、3 年と 4 年で 95% でした。血清テストステロンの増加の中央値は、1 年と 2 年で 19%、3 年で 26%、4 年で 22% でしたが、平均値と中央値は生理的範囲内にとどまりました。
経尿道的前立腺切除術の前にデュタステリドまたはプラセボを 1 日 5 mg 投与した BPH 患者では、デュタステリド群の前立腺組織の平均 DHT 濃度は、プラセボ群と比較して有意に低かった (784 および 5,793 pg/g)。 、それぞれ、P
遺伝的に継承された 2 型 5 アルファ還元酵素欠乏症の成人男性も、DHT レベルが低下しています。これらの 5 つのアルファ還元酵素欠損男性は、生涯を通じて前立腺が小さく、BPH を発症しません。出生時に存在する関連する泌尿生殖器の欠陥を除いて、5α-レダクターゼ欠損症に関連する他の臨床的異常はこれらの個人では観察されていません.
他のホルモンへの影響
健康なボランティアでは、デュタステリド 0.5 mg/日 (n = 26) による 52 週間の治療は、プラセボ (n = 23) と比較して、性ホルモン結合グロブリン、エストラジオール、黄体形成ホルモン、卵胞刺激ホルモン、チロキシン (遊離 T4)、およびデヒドロエピアンドロステロン。プラセボと比較して統計的に有意な、ベースライン調整後の平均増加が、8 週間で総テストステロン (97.1 ng/dL、P
その他の効果
血漿脂質パネルと骨ミネラル密度は、健康なボランティアにデュタステリド 0.5 mg を 1 日 1 回 52 週間投与した後に評価されました。プラセボまたはベースラインと比較して、二重エネルギーX線吸収法で測定した骨密度に変化はありませんでした。さらに、血漿脂質プロファイル (すなわち、総コレステロール、低密度リポタンパク質、高密度リポタンパク質、トリグリセリド) はデュタステリドの影響を受けませんでした。副腎皮質刺激ホルモン (ACTH) 刺激に対する副腎ホルモン応答の臨床的に有意な変化は、1 年間の健康なボランティア試験のサブセット集団 (n = 13) で観察されませんでした。
薬物動態
吸収
0.5 mg のソフト ゼラチン カプセルを 1 回投与した後、デュタステリドの血清中濃度がピークに達するまでの時間 (Tmax) は 2 ~ 3 時間以内です。 5 人の健常者における絶対バイオアベイラビリティは約 60% (範囲: 40% から 94%) です。この薬を食事と一緒に投与すると、最大血清濃度が 10% から 15% 低下しました。この減少は臨床的に重要ではありません。
分布
単回および反復経口投与後の薬物動態データは、デュタステリドが大量の分布 (300 ~ 500 L) を有することを示しています。デュタステリドは、血漿アルブミン (99.0%) およびα-1 酸性糖タンパク質 (96.6%) と高度に結合しています。
健康な被験者 (n = 26) がデュタステリド 0.5 mg/日を 12 か月間投与された試験では、精液デュタステリド濃度は 12 か月で平均 3.4 ng/mL (範囲: 0.4 から 14 ng/mL) であり、血清と同様に安定していました。 -6ヶ月での状態濃度。平均して、12 か月で血清デュタステリド濃度の 11.5% が精液に分配されました。
代謝と排泄
デュタステリドはヒトで広範囲に代謝されます。 In vitro 研究では、デュタステリドが CYP3A4 および CYP3A5 アイソザイムによって代謝されることが示されました。これらのアイソザイムは両方とも、4'-ヒドロキシデュタステリド、6-ヒドロキシデュタステリド、および 6,4'-ジヒドロキシデュタステリドの代謝産物を生成しました。さらに、15-ヒドロキシデュタステリド代謝産物は CYP3A4 によって形成されました。デュタステリドは、ヒト シトクロム P450 アイソザイム CYP1A2、CYP2A6、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6、および CYP2E1 によって in vitro で代謝されません。定常状態への投与後のヒト血清中、未変化のデュタステリド、3 つの主要代謝物 (4'-ヒドロキシデュタステリド、1,2-ジヒドロデュタステリド、および 6-ヒドロキシデュタステリド)、および 2 つの副代謝物 (6,4'-ジヒドロキシデュタステリドおよび 15 -ヒドロキシデュタステリド)、質量分析応答によって評価されるように、検出されました。 6 位と 15 位のヒドロキシル付加の絶対立体化学は不明です。インビトロでは、4'-ヒドロキシデュタステリドおよび1,2-ジヒドロデュタステリド代謝物は、ヒト5α-レダクターゼの両方のアイソフォームに対してデュタステリドよりもはるかに効力が低い. 6β-ヒドロキシデュタステリドの活性は、デュタステリドの活性に匹敵します。
デュタステリドとその代謝物は、主に糞便中に排泄されました。用量の割合として、変化していないデュタステリドが約 5% (約 1% から約 15%)、デュタステリド関連代謝物が約 40% (約 2% から約 90%) ありました。未変化のデュタステリドは微量しか尿中に検出されませんでした (
デュタステリドの最終消失半減期は、定常状態で約 5 週間です。平均定常状態の血清デュタステリド濃度は、0.5 mg/日を 1 年間投与した後、40 ng/mL でした。毎日の投薬後、デュタステリドの血清濃度は、1 か月後に定常状態の濃度の 65% に達し、3 か月後には約 90% に達します。デュタステリドの半減期が長いため、治療中止後も最大 4 ~ 6 か月間、血清濃度が検出可能 (0.1 ng/mL を超える) のままです。
特定の集団
小児患者
デュタステリドの薬物動態は、18 歳未満の被験者では調査されていません。
高齢患者
高齢者では用量調整は必要ありません。デュタステリドの薬物動態と薬力学は、デュタステリド 5 mg の単回投与後の 24 ~ 87 歳の 36 人の健康な男性被験者で評価されました。この単回投与試験では、デュタステリドの半減期は年齢とともに増加しました (20 歳から 49 歳の男性で約 170 時間、50 歳から 69 歳の男性で約 260 時間、70 歳以上の男性で約 300 時間)。 3 つのピボタル試験でデュタステリドの治療を受けた 2,167 人の男性のうち、60% が 65 歳以上、15% が 75 歳以上でした。これらの患者と若い患者の間で、安全性または有効性に全体的な違いは見られませんでした。
男性と女性の患者
AVODART は妊娠中は禁忌であり、女性への使用は適応されていません [参照 禁忌 、 警告と注意事項 ]。女性におけるデュタステリドの薬物動態は研究されていません。
人種および民族グループ
デュタステリドの薬物動態に対する人種の影響は研究されていません。
腎障害のある患者
デュタステリドの薬物動態に対する腎障害の影響は研究されていません。ただし、デュタステリドの定常状態の 0.5 mg 投与量の 0.1% 未満が人間の尿に回収されるため、腎障害のある患者の投与量の調整は予想されません。
肝障害のある患者
デュタステリドの薬物動態に対する肝障害の影響は研究されていません。デュタステリドは広範囲に代謝されるため、肝障害のある患者では曝露が高くなる可能性があります。
薬物相互作用研究
シトクロム P450 阻害剤
デュタステリドの薬物動態に対する CYP3A 酵素阻害剤の影響を評価するための臨床薬物相互作用試験は実施されていません。しかし、in vitro データに基づくと、リトナビル、ケトコナゾール、ベラパミル、ジルチアゼム、シメチジン、トロレアンドマイシン、シプロフロキサシンなどの CYP3A4/5 阻害剤の存在下で、デュタステリドの血中濃度が上昇する可能性があります。
デュタステリドは、1,000 ng/mL の濃度で主要なヒト シトクロム P450 アイソザイム (CYP1A2、CYP2C9、CYP2C19、CYP2D6、および CYP3A4) のモデル基質の in vitro 代謝を阻害しません。これは、ヒトの定常状態の血清濃度の 25 倍です。 .
α-アドレナリン拮抗薬
健康なボランティアを対象とした単一シーケンスのクロスオーバー試験では、AVODART と組み合わせたタムスロシンまたはテラゾシンの投与は、いずれのアルファアドレナリン拮抗薬の定常状態の薬物動態にも影響を与えませんでした。タムスロシンまたはテラゾシンの投与がデュタステリドの薬物動態パラメーターに及ぼす影響は評価されていませんが、DHT 濃度の変化率は、併用療法と比較して、AVODART 0.5mg 単独で同様でした。
カルシウムチャネル拮抗薬
集団薬物動態分析では、CYP3A4 阻害剤であるベラパミル (-37%、n = 6) およびジルチアゼム (-44%、n = 5) と併用した場合、デュタステリドのクリアランスの減少が認められました。対照的に、CYP3A4 阻害剤ではない別のカルシウム チャネル拮抗薬であるアムロジピンをデュタステリドと同時投与した場合、クリアランスの減少は見られませんでした (+7%、n = 4)。
ベラパミルとジルチアゼムの存在下でのクリアランスの減少とその後のデュタステリドへの暴露の増加は、臨床的に重要であるとは考えられていません.用量調整は推奨されません。
コレスチラミン
5 mg のアボダート 0.5 mg を単回投与し、1 時間後にコレスチラミン 12 g を投与しても、正常なボランティア 12 例におけるデュタステリドの相対的バイオアベイラビリティーに影響はありませんでした。
ジゴキシン
20 人の健康なボランティアを対象とした試験では、アボダート 0.5mg を 1 日 0.5mg の用量で 3 週間併用投与した場合、ジゴキシンの定常状態の薬物動態は変化しませんでした。
ワルファリン
23 人の健康なボランティアを対象とした試験では、1 日あたり 0.5 mg のアボダートを 3 週間投与しても、S-または R-ワルファリン異性体の定常状態の薬物動態は変化せず、ワルファリンと併用した場合のプロトロンビン時間に対するワルファリンの効果も変化しませんでした。
その他の併用療法
他の化合物との特定の相互作用試験は実施されていませんが、AVODART を投与された 3 つのランダム化二重盲検プラセボ対照安全性および有効性試験では、被験者の約 90% が他の薬剤を併用していました。 AVODART が抗高脂血症薬、アンギオテンシン変換酵素 (ACE) 阻害薬、ベータアドレナリン遮断薬、カルシウムチャネル遮断薬、コルチコステロイド、利尿薬、非ステロイド性抗-炎症薬(NSAID)、ホスホジエステラーゼ V 型阻害剤、およびキノロン系抗生物質。
動物毒物学および/または薬理学
中枢神経系毒性研究
ラットとイヌでは、デュタステリドを繰り返し経口投与した結果、何匹かの動物は、(親薬物の) 予想される臨床的曝露のそれぞれ 425 倍と 315 倍の曝露で、関連する組織病理学的変化を伴わない、非特異的で可逆的な中枢性毒性の徴候を示した。 .
ウサギの皮膚吸収
ウサギの皮膚薬物動態研究では、CAPMUL (オレイン酸グリセリル) 中のデュタステリドのウサギの皮膚吸収は、1 ~ 20 mg/mL の用量でそれぞれ 2.7 ~ 40.5 mcg/h/mL、または 56% ~ 100% の血清濃度をもたらしました。適用されたデュタステリドは、閉塞状態や長時間の条件下で吸収されます。経口投与されるアボダート ソフト ゼラチン カプセルには、カプリル酸/カプリン酸のモノジグリセリドとブチル化ヒドロキシトルエンの混合物に溶解した 0.5 mg のデュタステリドが含まれています。水中のデュタステリドは、ウサギに最小限しか吸収されませんでした (2,000 mg/kg)。
臨床研究
単剤療法
アボダート 0.5 mg/日 (n = 2,167) またはプラセボ (n = 2,158) は、3 つの 2 年間の多施設、プラセボ対照、二重盲検試験で BPH の男性被験者を対象に評価され、それぞれ 2 年間の非盲検延長 ( n = 2,340)。試験集団の 90% 以上が白人でした。被験者は少なくとも 50 歳で、血清 PSA が 1.5 ng/mL 以上 10 ng/mL 未満であり、BPH は病歴および身体検査によって診断され、前立腺肥大 (30 cc 以上) および中等度から重度の BPH 症状を含む。米国泌尿器科学会症状指数 (AUA-SI)。デュタステリドまたはプラセボのいずれかに無作為に割り当てられた 4,325 人の被験者のほとんどが、2 年間の二重盲検治療を完了しました (それぞれ 70% と 67%)。試験の延長に参加した 2,340 人の被験者のほとんどが、さらに 2 年間の非盲検治療を完了しました (71%)。
症状スコアへの影響
症状は、AUA-SI を使用して定量化されました。これは、尿の症状 (不完全な排尿、頻度、間欠性、切迫感、弱い尿流、いきみ、および夜間頻尿) を評価する質問票であり、0 から 5 のスケールで合計可能なスコア 35 で評価します。より高い数値の合計症状スコアは、症状の重症度が高いことを表しています。 3 つの試験のベースライン AUA-SI スコアは、両方の治療群で約 17 単位でした。
デュタステリドを投与された被験者は、1 つの試験では 3 か月まで、他の 2 つの重要な試験では 12 か月までに、プラセボに対して統計的に有意な症状の改善を達成しました。 12 か月目の時点で、プールされた 3 つの試験における AUA-SI の合計症状スコアのベースラインからの平均減少は、デュタステリドで -3.3 単位、プラセボで -2.0 単位であり、2 つの治療群の平均差は -1.3 (範囲: -1.1) でした。 3 回の試験のそれぞれで -1.5 単位、P
これらの試験は、ベースラインでの前立腺の大きさに基づいて症状への影響を評価するために前向きに設計されました。前立腺容積が 40 cc 以上の男性では、平均減少量はデュタステリドで -3.8 単位、プラセボで -1.6 単位であり、24 か月目の 2 つの治療群の平均差は -2.2 でした。前立腺容積が 40 cc 未満の男性では、平均減少は、デュタステリドで-3.7単位、プラセボで-2.2単位であり、24か月での2つの治療群の平均差は-1.5でした.
図 1: ベースラインからの AUA-SI スコアの変化 (無作為化、二重盲検、プラセボ対照試験をプール)
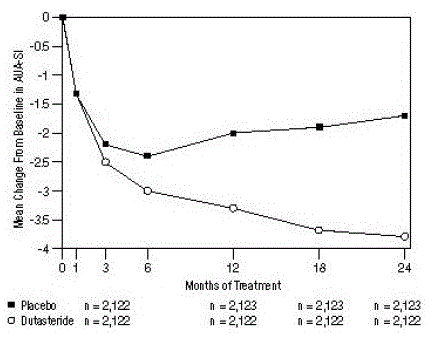
AUA-SI スコアの範囲は 0 ~ 35 です。
急性尿閉への影響とBPH関連の手術の必要性
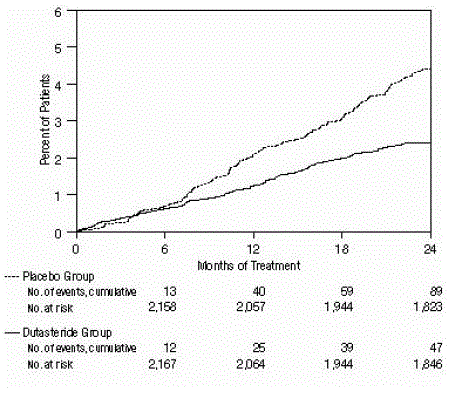
有効性は、2 年間の治療後に、カテーテル挿入および BPH 関連の泌尿器外科的介入を必要とする AUR の発生率によっても評価されました。プラセボと比較して、AVODART は統計的に有意に低い AUR の発生率と関連していました (AVODART で 1.8% 対 プラセボで 4.2%、P
図 2: 24 か月間に急性尿閉を発症した被験者の割合 (無作為化、二重盲検、プラセボ対照試験をプール)
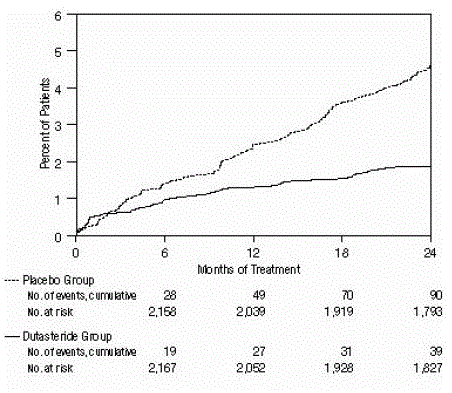
図 3: 24 か月間に良性前立腺肥大症の手術を受けた被験者の割合 (無作為化、二重盲検、プラセボ対照試験をプール)
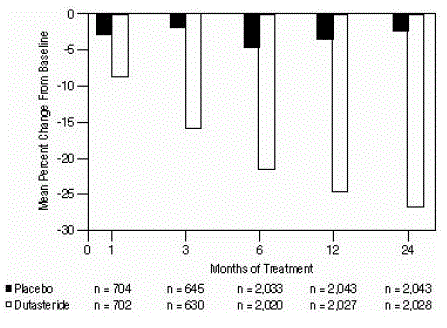
前立腺容積への影響
試験への参加には、経直腸超音波で測定した少なくとも 30 cc の前立腺容積が必要でした。試験参加時の平均前立腺容積は約 54 cc でした。
統計的に有意な差 (AVODART 0.5mg 対 プラセボ) は、各試験 (1 か月目、3 か月目、または 6 か月目) の治療後の最も早い前立腺容積測定で認められ、24 か月目まで続きました。プールされた3つの試験全体の前立腺容積は、デュタステリドで-24.7%、プラセボで-3.4%でした。平均差 (デュタステリドからプラセボを引いたもの) は -21.3% でした (範囲: 3 つの試験のそれぞれで -21.0% から -21.6%、P
図 4: ベースラインからの前立腺容積パーセント変化 (無作為化、二重盲検、プールされたプラセボ対照試験)
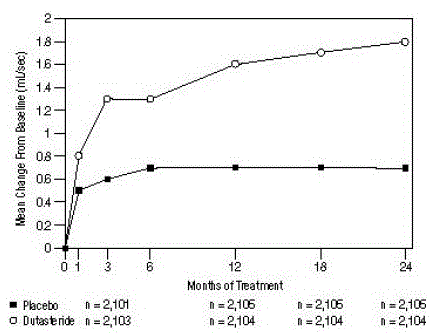
最大尿流量への影響
試験への参加には、15 mL/秒以下の平均ピーク尿流速 (Qmax) が必要でした。 Qmax は、3 つのピボタル試験全体でベースラインで約 10 mL/秒でした。
2 つのグループ間の差は、3 つの試験すべてで 3 か月目のベースラインから統計的に有意であり、12 か月目まで維持されました。プラセボの/秒。平均差 (デュタステリドからプラセボを差し引いた値) は 0.8 mL/秒でした (範囲: 3 つの試験のそれぞれで 0.7 ~ 1.0 mL/秒、P
図 5: ベースラインからの Qmax の変化 (プールされた無作為化、二重盲検、プラセボ対照試験)
臨床試験の概要
よく管理された 3 つの大規模な有効性試験のデータは、AVODART (0.5 mg 1 日 1 回) による治療が、プラセボと比較して AUR と BPH 関連の外科的介入の両方のリスクを減らし、BPH 関連の症状を改善し、前立腺容積を減少させ、最大値を増加させることを示しています。尿流量。これらのデータは、アボダート 0.5mg が前立腺肥大症の男性の BPH の疾患過程を阻止することを示唆しています。
α遮断薬療法(CombAT)との併用
併用療法 (アボダート 0.5 mg/日 + タムスロシン 0.4 mg/日、n = 1,610) の有効性を、アボダート 0.5 mg 単独 (n = 1,623) またはタムスロシン単独 (n = 1,611) と比較しました。 、二重盲検試験。試験への参加基準は、セクション 14.1 で説明した二重盲検プラセボ対照単剤療法の有効性試験と同様でした。登録された試験集団の 88% (88%) は白人でした。被験者の約 52% は、以前に 5 α 還元酵素阻害剤または α アドレナリン拮抗薬治療を受けたことがありました。無作為に割り付けられた 4,844 人の被験者のうち、併用群の被験者の 69%、アボダート 0.5mg の投与群の被験者の 67%、タムスロシン群の被験者の 61% が 4 年間の二重盲検治療を完了しました。
症状スコアへの影響
症状は、国際前立腺症状スコア (IPSS) (AUA-SI と同一) の最初の 7 つの質問を使用して定量化されました。ベースライン スコアは、各治療群で約 16.4 単位でした。併用療法は、このエンドポイントの主要な時点である 24 か月目の症状スコアの減少において、各単剤療法よりも統計的に優れていました。 24 か月目の IPSS の合計症状スコアのベースラインからの平均変化 (±SD) は、併用で -6.2 (±7.14)、AVODART で -4.9 (±6.81)、タムスロシンで -4.3 (±7.01) であり、平均差がありました。組み合わせとアボダート 0.5mg の間で -1.3 単位 (P
図 6: 48 か月間の国際前立腺症状スコアのベースラインからの変化 (無作為化、二重盲検、並行群間試験 [CombAT 試験])
![International Prostate Symptom Score Change from Baseline over a 48Month Period (Randomized, Double-blind, Parallel-Group Trial [CombAT Trial]) - Illustration](/pics/c721a1e7597b506f12d3ef49ee0f0482.jpg)
急性尿閉またはBPH関連手術の必要性への影響
年間の治療後、AVODART 0.5mg とタムスロシンの併用療法は、AUR または BPH 関連の手術の発生率を減少させる上で、AVODART の単独療法よりも有益ではありませんでした。
最大尿流量への影響
ベースラインの Qmax は、各治療群で約 10.7 mL/秒でした。併用療法は、このエンドポイントの主要な時点である 24 か月目の Qmax の増加において、各単剤療法よりも統計的に優れていました。 24 か月目の Qmax のベースラインからの平均増加量 (±SD) は、併用で 2.4 (±5.26) mL/秒、アボダート 0.5mg で 1.9 (±5.10) mL/秒、および併用で 0.9 (±4.57) mL/秒でした。タムスロシン、併用と AVODART の平均差は 0.5 mL/秒 (P = 0.003; [95% CI: 0.17, 0.84])、併用とタムスロシンの平均差は 1.5 mL/秒 (P
アボダート 0.5 mg の単剤療法に対する併用療法の Qmax の追加の改善は、48 か月目にはもはや統計的に有意ではありませんでした。
図 7: 24 か月間のベースラインからの Qmax の変化 (無作為化、二重盲検、並行群間試験 [CombAT 試験])
![Qmax Change from Baseline over a 24-Month Period (Randomized, Double-blind, Parallel-Group Trial [CombAT Trial]) - Illustration](/pics/903d9770522d9ac81293d6348b875483.jpg)
前立腺容積への影響
試験参加時の平均前立腺容積は約 55 cc でした。このエンドポイントの主要な時点である 24 か月目に、前立腺容積のベースラインからの平均変化率 (±SD) は、併用療法で -26.9% (±22.57)、アボダート 0.5mg で -28.0% (±24.88)、およびタムスロシンの場合は 0% (±31.14)、併用とアボダート 0.5mg の平均差は 1.1% (P = NS; [95% CI: -0.6, 2.8])、併用とタムスロシンの平均差は -26.9% (P
患者情報
アヴォダート (av o dart) (デュタステリド) カプセル
アボダート 0.5mg は男性専用です。
アボダート 0.5mg の服用を開始する前、およびリフィルを入手するたびに、この患者情報をお読みください。新しい情報があるかもしれません。この情報は、あなたの病状や治療について医療提供者と話すことに代わるものではありません.
アボダート0.5mgとは?
アボダート 0.5mg は、デュタステリドを含む処方薬です。アボダート 0.5mg は、前立腺肥大症の男性における良性前立腺肥大症 (BPH) の症状を治療するために使用されます。
- 症状を改善し、
- 急性尿閉(尿流の完全な閉塞)のリスクを軽減します。
- BPH 関連の手術が必要になるリスクを軽減します。
アボダートを服用してはいけない人
次の場合は、アボダートを服用しないでください。
- 妊娠している、または妊娠する可能性がある。アボダート 0.5mg は胎児に害を及ぼす可能性があります。妊娠中の女性はアボダート カプセルに触れないでください。男性の赤ちゃんを妊娠している女性が、アボダートを飲み込んだり触れたりして体内に十分な量のアボダート 0.5mg を摂取した場合、男性の赤ちゃんは正常でない性器を持って生まれてくる可能性があります。妊娠中の女性または出産の可能性のある女性が、漏れたアボダート 0.5mg カプセルに接触した場合は、接触部分を石鹸と水で直ちに洗い流してください。
- 子供またはティーンエイジャー。
- デュタステリドまたはアボダートの成分にアレルギーのある方。アボダートの成分の完全なリストについては、このリーフレットの最後を参照してください。
- PROSCAR(フィナステリド)®錠など、他の5α-レダクターゼ阻害剤にアレルギーのある方。
アボダート 0.5mg を服用する前に医療提供者に何を伝えるべきですか?
AVODART 0.5mg を服用する前に、次の場合は医療提供者に伝えてください。
- 肝臓に問題がある
処方薬、非処方薬、ビタミン、ハーブのサプリメントなど、服用しているすべての薬について医療提供者に伝えてください。アボダートと他の薬は互いに影響し合い、副作用を引き起こす可能性があります。 AVODART 0.5mg は他の医薬品の作用に影響を与える可能性があり、他の医薬品は AVODART 0.5mg の作用に影響を与える可能性があります。
服用している薬を知る。それらのリストを保管して、新しい薬を入手したときに医療提供者や薬剤師に見せてください.
アボダートはどのように服用すればよいですか?
- 1日1回1アボダートカプセルを服用してください。
- アボダート 0.5mg カプセルを丸ごと飲み込んでください。カプセルの内容物が唇、口、または喉を刺激する可能性があるため、アボダート 0.5mg カプセルをつぶしたり、噛んだり、開けたりしないでください。
- アボダート 0.5mg は食事の有無にかかわらず服用できます。
- 飲み忘れた場合は、その日のうちに服用することができます。翌日2回分を服用して、飲み忘れた分を補わないでください。
アボダート 0.5mg を服用している間、何を避けるべきですか?
- AVODART の服用中、または AVODART を中止してから 6 か月間は、献血を行うべきではありません。これは、妊婦が輸血によってアボダートを受けないようにするために重要です。
アボダートの副作用にはどのようなものがありますか?
AVODART は、次のような重大な副作用を引き起こす可能性があります。
- 以下を含むまれで深刻なアレルギー反応:
- 顔、舌、喉の腫れ
- 皮膚剥離などの深刻な皮膚反応
これらの深刻なアレルギー反応がある場合は、すぐに医師の診察を受けてください。
- より深刻な形態の前立腺がんの可能性が高くなります。
AVODART の最も一般的な副作用は次のとおりです。
- 勃起を得たり維持したりするのが困難(インポテンス)*
- 性欲(リビドー)の減少*
- 射精の問題*
- 乳房の肥大または痛み。乳房のしこりや乳頭分泌物に気付いた場合は、医療提供者に相談してください。
*これらのイベントの一部は、アボダートの服用を中止した後も続く場合があります.
AVODART を投与された患者では、気分の落ち込みが報告されています。
AVODART 0.5mg は、精子の数、精液の量、および精子の動きを減少させることが示されています。しかし、男性の生殖能力に対するアボダート 0.5mg の効果は知られていません。
前立腺特異抗原 (PSA) テスト: かかりつけの医療提供者は、アボダートの服用開始前および服用中に、前立腺がんなどの他の前立腺の問題についてチェックする場合があります。 PSA(前立腺特異抗原)と呼ばれる血液検査は、前立腺がんの可能性を確認するために使用されることがあります。 AVODART は、血液中の PSA 測定値を低下させます。あなたの医療提供者はこの影響を認識しており、PSAを使用して前立腺がんの可能性があるかどうかを確認できます. AVODART による治療中に PSA 値が上昇した場合 (PSA 値が正常範囲内であっても)、担当の医療提供者が評価する必要があります。
気になる副作用や治らない副作用がある場合は、医療提供者に伝えてください。
これらは、AVODART で考えられるすべての副作用ではありません。詳細については、医療提供者または薬剤師にお尋ねください。
副作用に関する医学的アドバイスについては、医師に連絡してください。 1-800-FDA-1088 で副作用を FDA に報告できます。
アボダート 0.5mg の保存方法を教えてください。
- アボダート 0.5mg カプセルは室温 (59°F ~ 86°F または 15°C ~ 30°C) で保管してください。
- アボダート 0.5mg カプセルは、高温で保管すると変形および/または変色することがあります。
- カプセルが変形、変色、または漏れている場合は、アボダート 0.5mg を使用しないでください。
- いらない薬は安全に捨てる。
アボダート 0.5mg およびすべての医薬品は、子供の手の届かないところに保管してください。
医薬品は、患者リーフレットに記載されている以外の目的で処方されることがあります。アボダート 0.5mg は、処方されていない状態には使用しないでください。他の人があなたと同じ症状を持っている場合でも、AVODART を与えないでください。それらに害を及ぼす可能性があります。
この患者情報リーフレットには、AVODART に関する最も重要な情報がまとめられています。さらに詳しい情報が必要な場合は、医療提供者に相談してください。医療専門家向けに書かれた AVODART 0.5mg に関する情報については、薬剤師または医療提供者にお尋ねください。
詳細については、www.AVODART.com にアクセスするか、1-888-825-5249 までお電話ください。
アボダート 0.5mg の成分は何ですか?
有効成分: デュタステリド。
不活性成分 : ブチル化ヒドロキシトルエン、酸化第二鉄(黄色)、ゼラチン(BSEフリーの認定牛源由来)、グリセリン、カプリル酸/カプリン酸のモノジグリセリド、二酸化チタン、食用赤インク。
アボダートはどのように機能しますか?
前立腺肥大は、ジヒドロテストステロン(DHT)と呼ばれる血液中のホルモンによって引き起こされます。 AVODART は体内の DHT 産生を低下させ、ほとんどの男性で肥大した前立腺を縮小させます。一部の男性では、アボダートによる 3 か月の治療後に問題や症状が軽減されますが、アボダート 0.5mg が効果的かどうかを確認するには、通常、少なくとも 6 か月の治療期間が必要です。
この患者情報は、米国食品医薬品局によって承認されています。