目薬: Lumigan 3ml 使用法、副作用および投与量。 オンライン薬局の価格。 処方箋不要のジェネリック医薬品。
ルミガンとは?
ルミガン 3ml は、特定のタイプの緑内障およびまつげの貧毛症によって引き起こされる眼圧上昇の症状を治療するために使用される処方薬です。ルミガンは、単独で使用することも、他の薬と併用することもできます。
ルミガンは、抗緑内障、プロスタグランジン作動薬と呼ばれる薬のクラスに属しています。
ルミガンが子供に安全で効果があるかどうかはわかっていません.
ルミガンの副作用は?
ルミガンは、次のような重大な副作用を引き起こす可能性があります。
- 目の腫れ、
- 目の充血、
- 目の重度の不快感、
- 目からの痂皮または排水、
- 視界が変わり、
- まぶたの赤み、腫れ、かゆみ
上記の症状がある場合は、すぐに医療機関を受診してください。
ルミガンの最も一般的な副作用は次のとおりです。
- 目の充血
気になる副作用や治らない副作用がある場合は、医師に相談してください。
ルミガンの副作用はこれだけではありません。詳細については、医師または薬剤師にお尋ねください。
副作用に関する医学的アドバイスについては、医師に連絡してください。 1-800-FDA-1088 で副作用を FDA に報告できます。
説明
LUMIGAN® (ビマトプロスト点眼液) 0.03% は、眼圧降下作用を持つ合成プロスタミド類似体です。その化学名は (Z)-7-[(1R,2R,3R,5S)-3,5-ジヒドロキシ-2[(1E,3S)-3-ヒドロキシ-5-フェニル-1-ペンテニル]シクロペンチル]- 5-N-エチルヘプテンアミドで、分子量は415.58です。その分子式は C25H37NO4 です。その化学構造は次のとおりです。
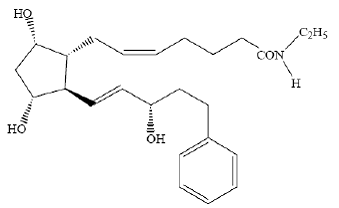
ビマトプロストは粉末で、エチルアルコールとメチルアルコールに非常に溶けやすく、水にはほとんど溶けません。 LUMIGAN® 0.03% は、浸透圧が約 290 mOsmol/kg の透明、等張性、無色、無菌の点眼液です。
ルミガン® 0.03%含有 アクティブ: ビマトプロスト 0.3 mg/mL; 非アクティブ: 塩化ベンザルコニウム 0.05 mg/mL;塩化ナトリウム;リン酸ナトリウム、二塩基性;クエン酸;そして精製水。水酸化ナトリウムおよび/または塩酸を添加してpHを調整することができる。保存期間中の pH は 6.8 ~ 7.8 の範囲です。
適応症
LUMIGAN® (ビマトプロスト点眼液) 0.01% は、開放隅角緑内障または高眼圧症患者の眼圧上昇の低減に適応されます。
投薬と管理
推奨用量は、1 日 1 回夕方に影響を受けた眼に 1 滴です。 LUMIGAN® (ビマトプロスト点眼液) 0.01% は、プロスタグランジン類似体をより頻繁に投与すると眼圧低下効果が低下する可能性があることが示されているため、1 日 1 回を超えて投与しないでください。
眼圧の低下は、最初の投与から約 4 時間後に始まり、約 8 ~ 12 時間以内に最大の効果に達します。
LUMIGAN® 0.01% は、眼圧を下げるために他の局所眼科用医薬品と併用することができます。複数の点眼薬を使用している場合、薬は少なくとも 5 分間隔で投与する必要があります。
供給方法
剤形と強度
ビマトプロスト 0.1 mg/mL を含む点眼液。
保管と取り扱い
LUMIGAN® (ビマトプロスト点眼液) 0.01% は、不透明な白色低密度ポリエチレン点眼用ディスペンサー ボトルとチップに無菌で供給され、以下のサイズのターコイズ ポリスチレン キャップが付いています。
2.5 mL を 5 mL 容器に充填 - NDC 0023-3205-03 10mL容器に5mL充填 - NDC 0023-3205-05 10mL容器に7.5mL充填 - NDC 0023-3205-08
保管所: 2°C から 25°C (36°F から 77°F) で保管してください。
LUMIGAN® 0.01%は開封後、ボトルに記載の使用期限までご使用いただけます。
配布元: Allergan USA, Inc. Madison, NJ 07940. 改訂: 2022 年 3 月
副作用
次の有害反応は、ラベルの別の場所に記載されています。
- 眼瞼色素沈着および虹彩過色素沈着を含む色素沈着[参照 警告と注意事項 ]
- まつ毛の変化 [参照] 警告と注意事項 ]
- 眼内炎症 [参照 警告と注意事項 ]
- 黄斑浮腫 [参照 警告と注意事項 ]
- 過敏症 [参照 禁忌 ]
臨床試験の経験
臨床試験はさまざまな条件下で実施されるため、ある医薬品の臨床試験で観察された副作用率を別の医薬品の臨床試験で観察された率と直接比較することはできず、実際に観察された率を反映していない可能性があります。
ビマトプロスト点眼液 0.01% を使用した 12 か月の臨床試験で、最も一般的な副作用は結膜充血 (31%) でした。患者の約 1.6% が結膜充血のために治療を中止しました。その他の副作用(患者の 1 ~ 4% で報告) ルミガン® この研究の 0.01% には、結膜浮腫、結膜出血、目の刺激、目の痛み、目のかゆみ、まぶたの紅斑、まぶたのかゆみ、まつげの成長、多毛症、点眼部位の刺激、点状角膜炎、皮膚の色素沈着、視力のぼやけ、および視力が含まれていました。減少。
市販後の経験
承認後の使用において、以下の副作用が確認されています。 ルミガン® 0.01%。これらの反応は不確かな規模の集団から自発的に報告されるため、その頻度を確実に推定したり、薬物曝露との因果関係を確立したりすることは常に可能ではありません.これらの反応は、その深刻さ、報告の頻度、および ルミガン® 、またはこれらの要因の組み合わせには、喘息様症状、めまい、ドライアイ、呼吸困難、眼脂、眼浮腫、異物感、頭痛、眼アレルギーおよびアレルギー性皮膚炎の徴候および症状を含む過敏症、高血圧、流涙の増加、まぶた溝の深化を含む眼窩周囲およびまぶたの変化、および羞明。
薬物相互作用
情報提供なし
警告
の一部として含まれています "予防" セクション
予防
色素沈着
ビマトプロスト点眼液は、色素組織に変化を引き起こすことが報告されています。最も頻繁に報告された変化は、虹彩、眼窩周囲組織 (まぶた) およびまつげの色素沈着の増加です。ビマトプロストが投与されている限り、色素沈着は増加すると予想されます。色素沈着の変化は、メラノサイトの数の増加ではなく、メラノサイトのメラニン含有量の増加によるものです。ビマトプロストの中止後、虹彩の色素沈着は永続的である可能性が高く、眼窩周囲組織の色素沈着およびまつげの変化は一部の患者で可逆的であると報告されています.治療を受ける患者には、色素沈着が増加する可能性があることを知らせる必要があります。色素沈着の長期的な影響は不明です。
虹彩の色の変化は、数か月から数年は目立たないことがあります。通常、瞳孔の周囲の茶色の色素沈着は、虹彩の周辺に向かって同心円状に広がり、虹彩全体または虹彩の一部がより茶色がかっていきます。虹彩の母斑もそばかすも治療による影響を受けないようです。ルミガン®(ビマトプロスト点眼液)0.01%での治療は、虹彩色素沈着が顕著に増加している患者に継続することができますが、これらの患者は定期的に検査する必要があります。
まつげの変化
LUMIGAN® 0.01% は、処理された目のまつ毛と軟毛を徐々に変化させる可能性があります。これらの変更には、まつげの長さ、太さ、および数の増加が含まれます。まつ毛の変化は通常、治療を中止すると元に戻ります。
眼内炎症
ビマトプロストを含むプロスタグランジン類似体は、眼内炎症を引き起こすことが報告されています。さらに、これらの製品は炎症を悪化させる可能性があるため、活動性の眼内炎症(ブドウ膜炎など)のある患者には注意が必要です。
黄斑浮腫
ビマトプロスト点眼液による治療中に、嚢胞様黄斑浮腫を含む黄斑浮腫が報告されています。 LUMIGAN® 0.01% は、無水晶体患者、後水晶体嚢が破れた偽水晶体患者、または黄斑浮腫の危険因子が知られている患者には注意して使用する必要があります。
細菌性角膜炎
局所眼科製品の複数回投与容器の使用に関連する細菌性角膜炎の報告があります。これらの容器は、ほとんどの場合、角膜疾患または眼上皮表面の破壊を併発している患者によって不注意に汚染されていました。
コンタクトレンズの使用
LUMIGAN® 0.01% には塩化ベンザルコニウムが含まれており、ソフトコンタクトレンズに吸収されて変色することがあります。コンタクト レンズは、LUMIGAN® 0.01% を点眼する前に取り外し、投与後 15 分後に再び挿入することができます。
非臨床毒性学
発がん、突然変異誘発、生殖能力の障害
発がん
ビマトプロストは、それぞれ最大 2 mg/kg/日および 1 mg/kg/日の用量で 104 週間強制経口投与された場合、マウスまたはラットのいずれにおいても発がん性がありませんでした (ビマトプロストへの推定ヒト全身曝露の 192 倍および 291 倍)。血中AUCレベルに基づいて、それぞれ1日1回両側投与)
突然変異誘発
ビマトプロストは、Ames 試験、マウスリンパ腫試験、または in vivo マウス小核試験において、変異原性または染色体異常誘発性を示さなかった。
生殖能力の障害
ビマトプロストは、0.6 mg/kg/日の用量まで雄または雌のラットの生殖能力を損なわなかった (血中 AUC レベルに基づいて、1 日 1 回両側に投与されるビマトプロスト 0.03% への推奨されるヒト曝露の少なくとも 103 倍)。
特定の集団での使用
妊娠
リスクの概要
LUMIGAN® (ビマトプロスト点眼液) 0.01% の妊娠中の女性への投与について、十分に管理された研究はありません。ビマトプロストの市販後の経験に基づくと、重大な先天性欠損症や流産のリスクが増加することはありません。
胚胎児発生研究では、器官形成中の妊娠中のマウスおよびラットへのビマトプロストの投与は、少なくとも 33 倍 (マウス) または 94 倍 (ラット) のビマトプロストへのヒト曝露で、1 日 1 回両側投与で流産および早期出産をもたらした (曲線下の血液面積 [AUC] レベルに基づく)。これらの悪影響は、ビマトプロスト 0.03% を 1 日 1 回両側投与したヒトの 2.6 倍 (マウス) および 47 倍 (ラット) では観察されませんでした (血中 AUC レベルに基づく)。
出生前/出生後の発育研究では、妊娠中のラットに器官形成から授乳の終わりまでビマトプロストを投与すると、妊娠期間と胎児の体重が減少し、ビマトプロストへのヒトの全身曝露の少なくとも 41 倍の経口投与で胎児と仔の死亡率が増加した 0.03 % 1 日 1 回両側投与 (血中 AUC レベルに基づく)。ビマトプロスト 0.03% を 1 日 1 回両側投与したヒトの曝露量の 14 倍と推定される曝露量では、ラットの子孫に悪影響は観察されませんでした (血中 AUC レベルに基づく)。
動物の生殖研究は、常にヒトの反応を予測できるとは限らないため、潜在的な利益が胎児への潜在的なリスクを正当化する場合にのみ、妊娠中に LUMIGAN® 0.01% を投与する必要があります。
データ
動物データ
胚胎児発生ラットの研究では、器官形成中にビマトプロストを 0.6 mg/kg/日で経口投与した妊娠ラットで流産が観察されました (AUC に基づいて、ビマトプロスト 0.03% を 1 日 1 回両側投与したヒトの全身暴露の 94 倍)。中絶の無毒性量 (NOAEL) は 0.3 mg/kg/日でした (AUC に基づいて、ビマトプロスト 0.03% を 1 日 1 回両側に投与するヒトの全身暴露量の 47 倍と推定)。 0.6 mg/kg/日までの用量でラット胎児に異常は観察されなかった。
胚胎児発生マウスの研究では、器官形成中にビマトプロストを 0.3 mg/kg/日以上の用量で経口投与された妊娠マウスで流産と早期出産が観察されました (ビマトプロスト 0.03% を 1 日 1 回両側投与したヒトの全身暴露の 33 倍、 AUCに基づく)。流産と早期分娩の NOAEL は 0.1 mg/kg/日でした (AUC に基づいて、ビマトプロスト 0.03% を 1 日 1 回両側投与したヒトの全身暴露の 2.6 倍)。 0.6 mg/kg/日までの用量でマウス胎児に異常は観察されなかった (AUC に基づいて、ビマトプロスト 0.03% を 1 日 1 回両側に投与したヒトの全身曝露の 72 倍)。
出生前/出生後の発育試験では、妊娠7日目から授乳20日目までビマトプロストを経口投与した妊娠ラットの治療により、妊娠期間が短縮され、後期吸収、胎児死亡、出生後の仔の死亡率が増加し、用量を超える用量で仔の体重が減少しました。または 0.3 mg/kg/日に等しい。これらの影響は、AUC に基づいて、1 日 1 回両側投与されたビマトプロスト 0.03% へのヒト全身暴露の少なくとも 41 倍の暴露で観察されました。子孫の出生後の発育および交配能力に関する NOAEL は 0.1 mg/kg/日であった (AUC に基づいて、ビマトプロスト 0.03% を 1 日 1 回両側に投与するヒトの全身暴露の 14 倍と推定)。
授乳
リスクの概要
LUMIGAN® 0.01% による局所眼科治療が、母乳で検出可能な量を生成するのに十分な全身吸収をもたらすかどうかは不明です。動物実験では、ビマトプロストは RHOD の 970 倍 (mg/rn 2 ベース) の静脈内投与量 (すなわち、1 mg/kg) で授乳中のラットの母乳に存在することが示されていますが、動物データは入手できません。臨床的に適切な用量で。
LUMIGAN® 0.01% に対する母親の臨床的必要性、および LUMIGAN® 0.01% による授乳中の子供への潜在的な悪影響とともに、母乳育児の発達上および健康上の利点を考慮する必要があります。
小児用
16 歳未満の小児患者への使用は、長期間の慢性使用による色素沈着の増加に関連する潜在的な安全上の懸念があるため、推奨されません。
高齢者の使用
高齢者とその他の成人患者との間で、安全性または有効性における全体的な臨床的差異は観察されていません。
過剰摂取
ヒトにおける過剰摂取に関する情報はありません。 LUMIGAN®(ビマトプロスト点眼液)0.01%の過剰摂取が発生した場合、治療は対症療法でなければなりません。
マウスとラットの経口 (強制経口投与による) 研究では、100 mg/kg/日までの用量で毒性は発生しませんでした。 mg/m2 で表されるこの用量は、10 kg の子供が LUMIGAN® 0.01% の 1 本の偶発的な用量よりも少なくとも 210 倍高いです。
禁忌
LUMIGAN® 0.01% は、ビマトプロストまたはいずれかの成分に対する過敏症の患者には禁忌です [参照 有害反応 ]。
臨床薬理学
作用機序
プロスタグランジン類似体であるビマトプロストは、プロスタグランジンの合成構造類似体であり、眼圧降下作用があります。天然物質であるプロスタミドの効果を選択的に模倣します。ビマトプロストは、小柱網とぶどう膜強膜経路の両方を介した房水の流出を増加させることにより、ヒトの眼圧 (IOP) を低下させると考えられています。上昇した IOP は、緑内障による視野損失の主要な危険因子を示します。 IOP のレベルが高いほど、視神経の損傷や視野喪失の可能性が高くなります。
薬物動態
吸収
ビマトプロスト点眼液 0.03% を 1 日 1 回、15 名の健常者の両眼に 1 滴 2 週間投与したところ、血中濃度は投与後 10 分以内にピークに達し、ほとんどの被験者で検出下限 (0.025 ng/mL) を下回りました。服用後1.5時間。 Cmax と AUC0-24hr の平均値は、7 日目と 14 日目でそれぞれ約 0.08 ng/mL と 0.09 ng·hr/mL で類似しており、眼内投与の最初の 1 週間で定常状態に達したことが示されました。時間の経過とともに有意な全身薬物蓄積はありませんでした。
分布
ビマトプロストは体組織に適度に分布し、定常状態の分布体積は 0.67 L/kg です。ヒトの血液では、ビマトプロストは主に血漿に存在します。ビマトプロストの約 12% は、ヒト血漿中で未結合のままです。
排除
代謝
ビマトプロストは、眼への投与後に体循環に到達すると、血液中の主要な循環種です。その後、ビマトプロストは酸化、N-脱エチル化、およびグルクロン酸抱合を受けて、多様な代謝産物を形成します。
排泄
放射性標識ビマトプロスト (3.12 mcg/kg) を 6 人の健康な被験者に静脈内投与した後、未変化体の最大血中濃度は 12.2 ng/mL であり、急速に減少し、消失半減期は約 45 分でした。ビマトプロストの総血中クリアランスは 1.5 L/hr/kg でした。投与量の最大 67% が尿中に排泄され、投与量の 25% が糞便中に回収されました。
臨床研究
平均ベースライン IOP が 23.5 mmHg の開放隅角緑内障または高眼圧症の患者を対象とした 12 か月間の臨床研究では、LUMIGAN® 0.01% を 1 日 1 回 (夕方) 投与した場合の眼圧低下効果は最大 7.5 mmHg でした。
患者情報
色素沈着の可能性
虹彩の茶色の色素沈着が増加する可能性があることを患者に伝えてください。これは永続的な可能性があります。また、LUMIGAN®(ビマトプロスト点眼液)0.01%の中止後に回復する可能性のあるまぶたの皮膚の黒ずみの可能性についても患者に知らせてください。
まつげの変化の可能性
LUMIGAN® 0.01% による治療中に、治療した眼のまつ毛と軟毛に変化が生じる可能性があることを患者に知らせてください。これらの変化は、長さ、太さ、色素沈着、まつげまたは軟毛の数、および/またはまつげの成長方向において、目の間に不均衡をもたらす可能性があります.まつ毛の変化は通常、治療を中止すると元に戻ります。
コンテナの取り扱い
眼の感染症を引き起こすことが知られている一般的な細菌による溶液の汚染を避けるために、調剤容器の先端が目、周囲の構造、指、またはその他の表面に触れないように患者に指示します。汚染された溶液を使用すると、眼に深刻な損傷を与え、視力を失う可能性があります。
いつ医師のアドバイスを求めるか
併発性眼疾患(外傷や感染症など)を発症した場合、眼科手術を受けた場合、または何らかの眼反応、特に結膜炎やまぶたの反応を発症した場合は、LUMIGAN® 0.01% の継続使用に関して直ちに医師の助言を求める必要があることを患者にアドバイスします。 .
コンタクトレンズの使用
LUMIGAN® 0.01% には塩化ベンザルコニウムが含まれており、ソフト コンタクト レンズに吸収される可能性があることを患者に伝えてください。コンタクト レンズは、LUMIGAN® 0.01% を点眼する前に取り外し、投与後 15 分後に再び挿入することができます。
他の眼科用薬との併用
複数の局所眼科用薬を使用している場合は、塗布の間隔を少なくとも 5 分空ける必要があることを患者に伝えてください。